



【New England Journal of Medicine】2025年6月-2025年7月刊论文导读
期刊介绍:
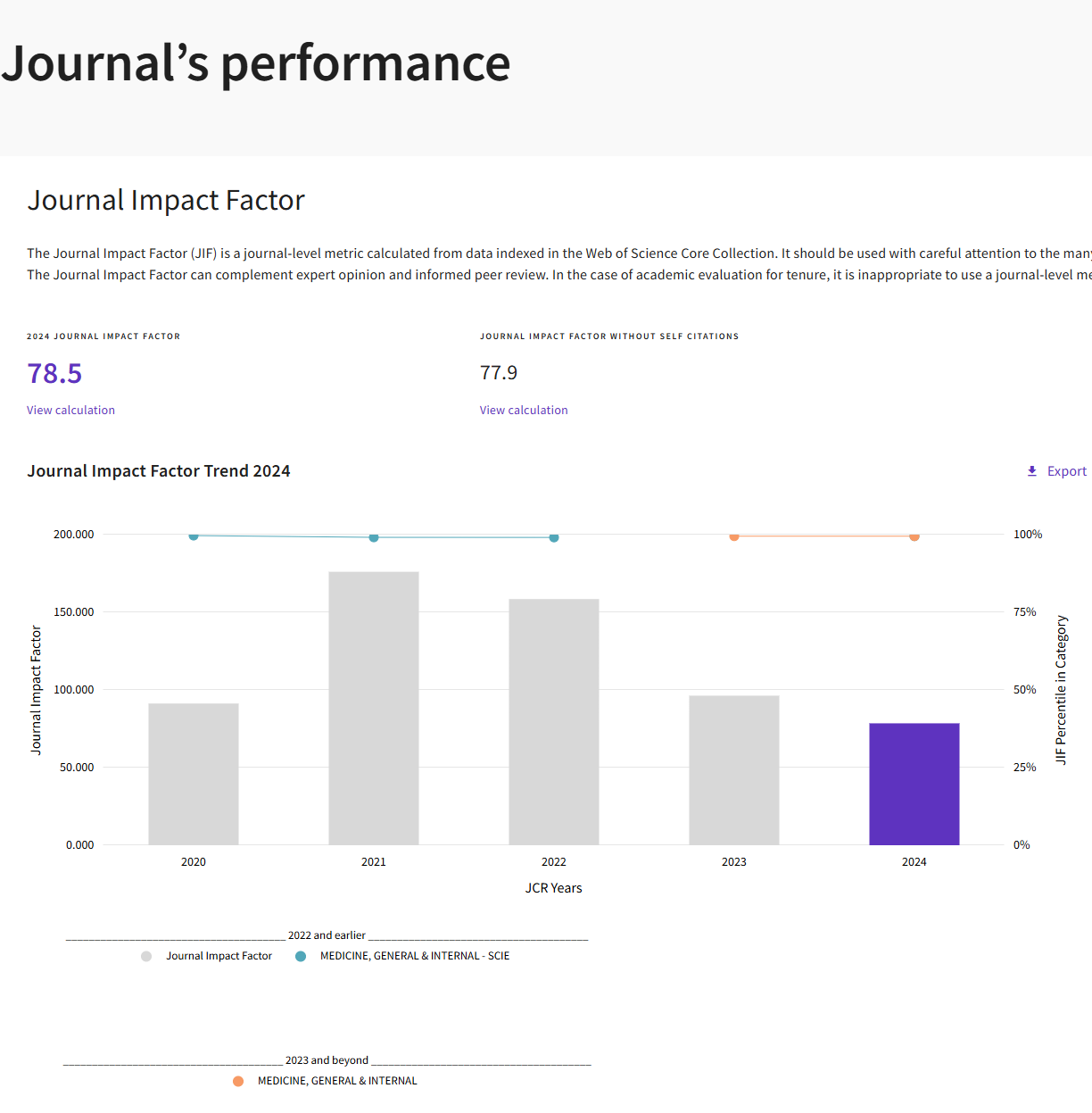
《新英格兰医学杂志》(New England Journal of Medicine, NEJM) 作为全球历史最悠久、影响力最卓著的综合性医学周刊,致力于发表最前沿、最攸关临床实践的重大医学研究成果。其内容深度融合原创性研究、大型临床试验与权威评论,旨在迅速将科学发现转化为临床诊疗指南,直接塑造全球医疗标准。其实2024年影响因子为78.5,被广泛视为临床医学领域的黄金标准与行业标杆。
New England Journal of Medicine
Volume 392(24) pgs. 2393-2496,e55-e56 June 26, 2025
ISSN: 0028-4793
Copyright (C) 2025 Massachusetts Medical Society. All rights reserved.
第392卷第24期一共发表34篇内容,其中原始研究3篇。
Routine Cerebral Embolic Protection during Transcatheter Aortic-Valve Implantation
经导管主动脉瓣植入术中的常规脑栓塞保护

牛津大学等机构联合发表
Abstract
Background: Transcatheter aortic-valve implantation (TAVI) is associated with procedure-related stroke. Cerebral embolic protection (CEP) devices may reduce embolization to the cerebral circulation and hence the incidence of stroke.
Methods: We conducted a randomized, controlled trial across 33 centers in the United Kingdom. We randomly assigned 7635 participants with aortic stenosis in a 1:1 ratio to undergo TAVI with a CEP device (CEP group) or TAVI without a CEP device (control group). The primary outcome was stroke within 72 hours after TAVI or before discharge from the hospital (if discharge occurred sooner).
Results: A total of 3815 participants were assigned to the CEP group and 3820 to the control group. A primary-outcome event occurred in 81 of 3795 participants (2.1%) in the CEP group and in 82 of 3799 participants (2.2%) in the control group (difference, -0.02 percentage points; 95% confidence interval, -0.68 to 0.63; P=0.94). Disabling stroke occurred in 47 participants (1.2%) in the CEP group and in 53 (1.4%) in the control group. Death occurred in 29 participants (0.8%) in the CEP group and in 26 (0.7%) in the control group. Overall access-site complications appeared to be similar in the two groups (8.1% in the CEP group and 7.7% in the control group). A total of 24 serious adverse events occurred in 22 of 3798 participants (0.6%) in the CEP group, and 13 serious adverse events occurred in 13 of 3803 participants (0.3%) in the control group.
Conclusions: Among participants undergoing TAVI, routine use of CEP did not decrease the incidence of stroke within 72 hours. (Funded by the British Heart Foundation and Boston Scientific; BHF PROTECT-TAVI ISRCTN Registry number, ISRCTN16665769.)
摘要
背景:经导管主动脉瓣植入术(TAVI)与手术相关的中风有关。脑栓塞保护(CEP)装置可能可减少栓子进入脑循环,从而降低中风的发生率。
方法:我们在英国的33个中心开展了一项随机对照试验。我们将7635名主动脉瓣狭窄患者以1:1的比例随机分配到使用CEP装置进行TAVI的组(CEP组)或不使用CEP装置进行TAVI的组(对照组)。主要终点是TAVI术后72小时内或出院前(以较早者为准)发生的中风。
结果:共有3815名参与者被分配到CEP组,3820名被分配到对照组。CEP组3795名参与者中有81人(2.1%)发生了主要终点事件,对照组3799名参与者中有82人(2.2%)发生主要终点事件(差异,-0.02个百分点;95%置信区间,-0.68至0.63;P=0.94)。CEP组47名参与者(1.2%)和对照组53名参与者(1.4%)发生了致残性中风。CEP组29名参与者(0.8%)和对照组26名参与者(0.7%)发生了死亡事件。两组的总体入路部位并发症相似(CEP组为8.1%,对照组为7.7%)。CEP组3798名参与者中有22人发生了24起严重不良事件(0.6%),对照组3803名参与者中有13人发生了13起严重不良事件(0.3%)。
结论:在进行TAVI的患者中,常规使用CEP并未降低术后72小时内中风的发生率。(由英国心脏基金会和波士顿科学公司资助;BHF PROTECT-TAVI ISRCTN注册号为ISRCTN16665769。)
Efruxifermin in Compensated Liver Cirrhosis Caused by MASH
Efruxifermin在代谢功能障碍相关脂肪性肝炎引起的代偿性肝硬化中的应用
美国休斯顿卫理公会医院肝病科及休斯顿研究所等机构联合发表
Abstract
Background: In phase 2 trials involving patients with stage 2 or 3 fibrosis caused by metabolic dysfunction-associated steatohepatitis (MASH), efruxifermin, a bivalent fibroblast growth factor 21 (FGF21) analogue, reduced fibrosis and resolved MASH. Data are needed on the efficacy and safety of efruxifermin in patients with compensated cirrhosis (stage 4 fibrosis) caused by MASH.
Methods: In this phase 2b, randomized, placebo-controlled, double-blind trial, we assigned patients with MASH who had biopsy-confirmed compensated cirrhosis (stage 4 fibrosis) to receive subcutaneous efruxifermin (at a dose of 28 mg or 50 mg once weekly) or placebo. The primary outcome was a reduction of at least one stage of fibrosis without worsening of MASH at week 36. Secondary outcomes included the same criterion at week 96.
Results: A total of 181 patients underwent randomization and received at least one dose of efruxifermin or placebo. Of these patients, liver biopsy was performed in 154 patients at 36 weeks and in 134 patients at 96 weeks. At 36 weeks, a reduction in fibrosis without worsening of MASH occurred in 8 of 61 patients (13%) in the placebo group, in 10 of 57 patients (18%) in the 28-mg efruxifermin group (difference from placebo after adjustment for stratification factors, 3 percentage points; 95% confidence interval [CI], -11 to 17; P=0.62), and in 12 of 63 patients (19%) in the 50-mg efruxifermin group (difference from placebo, 4 percentage points; 95% CI, -10 to 18; P=0.52). At week 96, a reduction in fibrosis without worsening of MASH occurred in 7 of 61 patients (11%) in the placebo group, in 12 of 57 patients (21%) in the 28-mg efruxifermin group (difference from placebo, 10 percentage points; 95% CI, -4 to 24), and in 18 of 63 patients (29%) in the 50-mg efruxifermin group (difference from placebo, 16 percentage points; 95% CI, 2 to 30). Gastrointestinal adverse events were more common with efruxifermin; most events were mild or moderate.
Conclusions: In patients with compensated cirrhosis caused by MASH, efruxifermin did not significantly reduce fibrosis at 36 weeks. (Funded by Akero Therapeutics; SYMMETRY ClinicalTrials.gov number, NCT05039450.)
摘要
背景:在针对代谢功能障碍相关脂肪性肝炎(MASH)所致2期或3期纤维化患者的II期临床试验中,双价成纤维细胞生长因子21(FGF21)类似物efruxifermin可减少肝纤维化并促使MASH缓解。然而,尚缺乏其在由MASH所致代偿性肝硬化(4期纤维化)患者中的疗效与安全性数据。
方法:在这项 IIb 期、随机、安慰剂对照、双盲试验中,我们将经活检证实的肝硬化(4 期纤维化)的 MASH 患者分配到皮下注射依鲁昔芬明(每周一次,剂量为 28 mg 或 50 mg)或安慰剂组。主要疗效指标为第 36 周时纤维化至少改善一个分期且 MASH 未加重。次要疗效指标为第 96 周时相同的标准。
结果:共有 181 名患者接受了随机分组,并接受了至少一剂依鲁昔芬明或安慰剂。在这些患者中,36 周时对 154 名患者进行了肝活检,96 周时对 134 名患者进行了肝活检。在第 36 周,安慰剂组中有 61 名患者(13%)出现纤维化减轻且 MASH 未加重,28 mg 依鲁昔芬明组中有 57 名患者(18%)出现(经分层因素调整后的与安慰剂的差异为 3 个百分点;95% 置信区间 [CI],-11 至 17;P=0.62),50 mg 依鲁昔芬明组中有 63 名患者(19%)出现(与安慰剂的差异为 4 个百分点;95% CI,-10 至 18;P=0.52)。在第 96 周,安慰剂组中有 61 名患者(11%)出现纤维化减轻且 MASH 未加重,28 mg 依鲁昔芬明组中有 57 名患者(21%)出现(与安慰剂的差异为 10 个百分点;95% CI,-4 至 24),50 mg 依鲁昔芬明组中有 63 名患者(29%)出现(与安慰剂的差异为 16 个百分点;95% CI,2 至 30)。胃肠道不良事件在使用依鲁昔芬明组中更常见;大多数事件为轻度或中度。
结论:在由 MASH 引起的失代偿期肝硬化患者中,依鲁昔芬明在 36 周时并未显著减轻纤维化。(由 Akero Therapeutics 资助;SYMMETRY 临床试验注册号 NCT05039450。)
Encorafenib, Cetuximab, and mFOLFOX6 in BRAF-Mutated Colorectal Cancer
恩曲非尼、西妥昔单抗和mFOLFOX6在BRAF突变型结直肠癌中的应用

西班牙巴塞罗那Vall d’Hebron医院等机构联合发表
Abstract
Background: First-line treatment with encorafenib plus cetuximab (EC) with or without chemotherapy (oxaliplatin, leucovorin, and fluorouracil [mFOLFOX6]) for BRAF V600E-mutated metastatic colorectal cancer, an aggressive subtype with a poor prognosis, was compared with standard care (chemotherapy with or without bevacizumab) in an open-label, phase 3 trial, which showed significance regarding one of the two primary end points, objective response according to blinded independent central review (odds ratio for EC+mFOLFOX6 vs. standard care, 2.44; one-sided P<0.001). This result led to accelerated Food and Drug Administration approval of this investigational combination therapy for BRAF V600E-mutated metastatic colorectal cancer, including as first-line therapy. Data on progression-free survival (the second primary end point) and an updated interim analysis of overall survival are now available.
Methods: We randomly assigned patients with untreated BRAF V600E-mutated metastatic colorectal cancer to receive EC, EC+mFOLFOX6, or standard care. The two primary end points were objective response (reported previously) and progression-free survival according to blinded independent central review in the EC+mFOLFOX6 group and the standard-care group. The key secondary end point was overall survival.
Results: Significantly longer progression-free survival was seen with EC+mFOLFOX6 than with standard care (median, 12.8 vs. 7.1 months; hazard ratio for progression or death, 0.53; 95% confidence interval [CI], 0.41 to 0.68; P<0.001). In an interim analysis, overall survival was significantly longer with EC+mFOLFOX6 than with standard care (median, 30.3 vs. 15.1 months; hazard ratio for death, 0.49; 95% CI, 0.38 to 0.63; P<0.001). The incidence of serious adverse events during treatment was 46.1% with EC+mFOLFOX6 and 38.9% with standard care. Safety profiles were consistent with those known for each agent.
Conclusions: This trial showed significantly longer progression-free survival and overall survival with first-line treatment with EC+mFOLFOX6 than with standard care among patients with BRAF V600E-mutated metastatic colorectal cancer. (Funded by Pfizer and others; BREAKWATER ClinicalTrials.gov number, NCT04607421.)
摘要
背景:在一项开放标签的 III 期试验中,将恩科拉芬尼联合西妥昔单抗(EC)联合或不联合化疗(奥沙利铂、亚叶酸、氟尿嘧啶 [mFOLFOX6])作为 BRAF V600E 突变转移性结直肠癌(一种预后不良的侵袭性亚型)的一线治疗,与标准治疗(化疗联合或不联合贝伐珠单抗)进行了比较。该试验在两个主要终点之一——根据盲法独立中心评审的客观缓解率方面显示出显著性(EC+mFOLFOX6 与标准治疗的优势比为 2.44;单侧 P<0.001)。这一结果促使美国食品药品监督管理局(FDA)加速批准了该研究性联合疗法用于 BRAF V600E 突变转移性结直肠癌,包括作为一线治疗。关于无进展生存期(第二个主要终点)的数据以及总生存期更新的期中分析现已可用。
方法:我们将未经治疗的BRAF V600E突变型转移性结直肠癌患者随机分配至接受EC、EC+mFOLFOX6或标准治疗组。两个主要终点分别为客观反应(此前已报告)以及由盲法独立中心审查评估的EC+mFOLFOX6组与标准治疗组的无进展生存期。关键次要终点为总生存期。
结果:在中期分析中,EC+mFOLFOX6组的总生存期也显著长于标准治疗组(中位数分别为30.3个月与15.1个月;死亡风险比为0.49;95% CI为0.38~0.63;P<0.001)。在治疗期间,EC+mFOLFOX6组发生严重不良事件的比例为46.1%,标准治疗组为38.9%。各治疗方案的安全性特征与已知情况一致。
结论:本试验表明,在BRAF V600E突变型转移性结直肠癌患者中,一线治疗使用EC+mFOLFOX6方案的无进展生存期和总生存期均显著长于标准治疗方案。(由辉瑞公司等资助;BREAKWATER试验ClinicalTrials.gov注册号为NCT04607421。)
第393卷第1期一共发表33,其中原始研究4篇。
Structured Exercise after Adjuvant Chemotherapy for Colon Cancer针对接受辅助化疗后的结肠癌患者开展的结构化运动方案 
加拿大癌症临床试验组等机构联合发表
Abstract
Background: Preclinical and observational studies suggest that exercise may improve cancer outcomes. However, definitive level 1 evidence is lacking.
Methods: In this phase 3, randomized trial conducted at 55 centers, we assigned patients with resected colon cancer who had completed adjuvant chemotherapy to participate in a structured exercise program (exercise group) or to receive health-education materials alone (health-education group) over a 3-year period. The primary end point was disease-free survival.
Results: From 2009 through 2024, a total of 889 patients underwent randomization to the exercise group (445 patients) or the health-education group (444 patients). At a median follow-up of 7.9 years, disease-free survival was significantly longer in the exercise group than in the health-education group (hazard ratio for disease recurrence, new primary cancer, or death, 0.72; 95% confidence interval [CI], 0.55 to 0.94; P=0.02). The 5-year disease-free survival was 80.3% in the exercise group and 73.9% in the health-education group (difference, 6.4 percentage points; 95% CI, 0.6 to 12.2). Results support longer overall survival in the exercise group than in the health-education group (hazard ratio for death, 0.63; 95% CI, 0.43 to 0.94). The 8-year overall survival was 90.3% in the exercise group and 83.2% in the health-education group (difference, 7.1 percentage points; 95% CI, 1.8 to 12.3). Musculoskeletal adverse events occurred more often in the exercise group than in the health-education group (in 18.5% vs. 11.5% of patients).
Conclusions: A 3-year structured exercise program initiated soon after adjuvant chemotherapy for colon cancer resulted in significantly longer disease-free survival and findings consistent with longer overall survival. (Funded by the Canadian Cancer Society and others; CHALLENGE ClinicalTrials.gov number, NCT00819208.)
摘要
背景:临床前和观察性研究表明,运动可能改善癌症的预后。然而目前仍缺乏确凿的一级证据。
方法:在这项55个中心进行的III期随机对照试验中,我们将已接受结肠癌手术切除并完成辅助化疗的患者随机分配至结构化运动计划组(运动组)或仅接受健康教育资料的对照组(健康教育组),研究周期为3年。主要终点为无病生存期。
结果:从2009年到2024年,共有889名患者被随机分配到运动组(445名患者)或健康教育组(444名患者)。在中位随访7.9年的情况下,运动组的无病生存期显著长于健康教育组(疾病复发、新发原发癌或死亡的风险比为0.72;95%置信区间[CI],0.55至0.94;P=0.02)。运动组的5年无病生存率为80.3%,健康教育组为73.9%(差异为6.4个百分点;95% CI,0.6至12.2)。运动组的总生存期也明显长于健康教育组(死亡的风险比为0.63;95% CI,0.43至0.94)。运动组的8年总生存率为90.3%,健康教育组为83.2%(差异为7.1个百分点;95% CI,1.8至12.3)。运动组的肌肉骨骼不良事件发生率高于健康教育组(分别为18.5%与11.5%)。
结论:一项在结肠癌辅助化疗后不久启动的为期3年的结构化运动计划,显著延长了无病生存期,并发现了与更长的总生存期一致的结果。(由加拿大癌症协会等机构资助;CHALLENGE ClinicalTrials.gov 注册号,NCT00819208。)
Tirzepatide as Compared with Semaglutide for the Treatment of Obesity
替尔泊肽与司美格鲁肽在肥胖治疗中的比较研究
美国得克萨斯大学等机构联合发表
Abstract
Background: Tirzepatide and semaglutide are highly effective medications for obesity management. The efficacy and safety of tirzepatide as compared with semaglutide in adults with obesity but without type 2 diabetes is unknown.
Methods: In this phase 3b, open-label, controlled trial, adult participants with obesity but without type 2 diabetes were randomly assigned in a 1:1 ratio to receive the maximum tolerated dose of tirzepatide (10 mg or 15 mg) or the maximum tolerated dose of semaglutide (1.7 mg or 2.4 mg) subcutaneously once weekly for 72 weeks. The primary end point was the percent change in weight from baseline to week 72. Key secondary end points included weight reductions of at least 10%, 15%, 20%, and 25% and a change in waist circumference from baseline to week 72.
Results: A total of 751 participants underwent randomization. The least-squares mean percent change in weight at week 72 was -20.2% (95% confidence interval [CI], -21.4 to -19.1) with tirzepatide and -13.7% (95% CI, -14.9 to -12.6) with semaglutide (P<0.001). The least-squares mean change in waist circumference was -18.4 cm (95% CI, -19.6 to -17.2) with tirzepatide and -13.0 cm (95% CI, -14.3 to -11.7) with semaglutide (P<0.001). Participants in the tirzepatide group were more likely than those in the semaglutide group to have weight reductions of at least 10%, 15%, 20%, and 25%. The most common adverse events in both treatment groups were gastrointestinal, and most were mild to moderate in severity and occurred primarily during dose escalation.
Conclusions: Among participants with obesity but without diabetes, treatment with tirzepatide was superior to treatment with semaglutide with respect to reduction in body weight and waist circumference at week 72. (Funded by Eli Lilly; SURMOUNT-5 ClinicalTrials.gov number, NCT05822830.)
摘要
背景: Tirzepatide和Semaglutide是两种在肥胖管理中非常有效的药物。对于没有2型糖尿病的肥胖成年人,Tirzepatide与Semaglutide的疗效和安全性尚不明确。
方法: 在这项 IIIb 期、开放标签、对照研究中,肥胖但无2型糖尿病的成年参与者被按1:1的比例随机分配,接受 tirzepatide 的最大耐受剂量(10 mg 或 15 mg)或 semaglutide 的最大耐受剂量(1.7 mg 或 2.4 mg),每周皮下注射一次,持续72周。主要终点为基线至第72周的体重百分比变化。关键次要终点包括体重至少减少10%、15%、20%、和25%的比例,以及基线至第72周腰围的变化。
结果: 共751名参与者接受随机分组。第72周时,替西帕肽组的体重最小二乘平均变化百分比为-20.2%(95%置信区间[CI]为-21.4至-19.1),司美格鲁肽组为-13.7%(95% CI为-14.9至-12.6)(P<0.001)。替西帕肽组的腰围最小二乘平均变化为-18.4厘米(95% CI为-19.6至-17.2),司美格鲁肽组为-13.0厘米(95% CI为-14.3至-11.7)(P<0.001)。与司美格鲁肽组相比,替西帕肽组参与者实现体重下降至少10%、15%、20%和25%的比例更高。两组最常见的不良事件均为胃肠道反应,多数严重程度为轻至中度,且主要发生在剂量递增期间。
结论: 在患有肥胖症但无糖尿病的参与者中,替西帕肽治疗在72周时对体重和腰围的改善效果优于司美格鲁肽。(资助单位:Eli Lilly;SURMOUNT-5 临床试验编号:NCT05822830。)
Neoadjuvant and Adjuvant Pembrolizumab in Locally Advanced Head and Neck Cancer
局部晚期头颈部癌的新辅助与辅助帕博利珠单抗治疗
美国哈佛医学院附属布莱根妇女医院等机构联合发表
Abstract
Background: The benefit of the addition of perioperative pembrolizumab to standard care with surgery and adjuvant therapy for patients with locally advanced head and neck squamous-cell carcinoma (HNSCC) is unclear.
Methods: In this phase 3, open-label trial, we randomly assigned participants with locally advanced HNSCC in a 1:1 ratio to receive 2 cycles of neoadjuvant pembrolizumab and 15 cycles of adjuvant pembrolizumab (both at a dose of 200 mg every 3 weeks) in addition to standard care (pembrolizumab group) or standard care alone (control group). Standard care was surgery and adjuvant radiotherapy with or without concomitant cisplatin. The primary end point was event-free survival, sequentially assessed in participants whose tumors expressed programmed death ligand 1 (PD-L1) with a combined positive score (CPS) of 10 or more (CPS-10 population), participants whose tumors expressed PD-L1 with a CPS of 1 or more (CPS-1 population), and all the participants. A higher CPS indicates a higher proportion of cells that express PD-L1.
Results: A total of 363 participants (234 with a CPS of >=10 and 347 with a CPS of >=1) were assigned to the pembrolizumab group and 351 (231 with a CPS of >=10 and 335 with a CPS of >=1) to the control group. Surgery was completed in approximately 88% of the participants in each group. At the first interim analysis, the median follow-up was 38.3 months. Event-free survival at 36 months was 59.8% in the pembrolizumab group and 45.9% in the control group (hazard ratio for progression, recurrence, or death, 0.66; 95% confidence interval [CI], 0.49 to 0.88; two-sided P=0.004) in the CPS-10 population; 58.2% and 44.9%, respectively (hazard ratio, 0.70; 95% CI, 0.55 to 0.89; two-sided P=0.003), in the CPS-1 population; and 57.6% and 46.4%, respectively (hazard ratio, 0.73; 95% CI, 0.58 to 0.92; two-sided P=0.008), in the total population. Grade 3 or higher treatment-related adverse events occurred in 44.6% of the participants in the pembrolizumab group and in 42.9% of those in the control group, including death in 1.1% and 0.3%, respectively. Potentially immune-mediated adverse events of grade 3 or higher occurred in 10.0% of the participants in the pembrolizumab group.
Conclusions: The addition of neoadjuvant and adjuvant pembrolizumab to standard care significantly improved event-free survival among participants with locally advanced HNSCC. Neoadjuvant pembrolizumab did not affect the likelihood of surgical completion. No new safety signals were identified. (Funded by Merck Sharp and Dohme, a subsidiary of Merck [Rahway, NJ]; KEYNOTE-689 ClinicalTrials.gov number, NCT03765918.)
摘要
背景:在局部晚期头颈部鳞状细胞癌(HNSCC)患者的治疗中,术前和术后使用帕博利珠单抗联合标准治疗(包括手术和辅助治疗)的益处尚不明确。
方法:在这项第三阶段、开放标签的随机对照试验中,我们将局部晚期HNSCC的参与者以1:1的比例随机分配至两组:一组接受2个周期的新辅助帕博利珠单抗和15个周期的辅助帕博利珠单抗(每3周200 mg),并结合标准治疗(帕博利珠单抗组);另一组仅接受标准治疗(对照组)。标准治疗包括手术和辅助放疗,可选择性使用顺铂。主要终点为事件无进展生存期,分别在以下三组人群中进行评估:肿瘤表达程序性死亡配体1(PD-L1)且合并阳性评分(CPS)≥10的患者(CPS-10组),肿瘤表达PD-L1且CPS≥1的患者(CPS-1组),以及所有参与者。CPS越高,表示PD-L1表达的细胞比例越高。
结果:共有363名参与者(其中234人CPS≥10,347人CPS≥1)被分配到帕博利珠单抗组,351名参与者(其中231人CPS≥10,335人CPS≥1)被分配到对照组。每组约88%的参与者完成了手术。在第一次中期分析时,随访的中位时间为38.3个月。在36个月时,帕博利珠单抗组的事件无进展生存率为59.8%,对照组为45.9%(CPS-10组,进展、复发或死亡的风险比为0.66,95%置信区间[CI]:0.49至0.88,双侧P=0.004);CPS-1组分别为58.2%和44.9%(风险比0.70,95% CI:0.55至0.89,双侧P=0.003);总体人群分别为57.6%和46.4%(风险比0.73,95% CI:0.58至0.92,双侧P=0.008)。帕博利珠单抗组中,3级及以上治疗相关的不良事件发生率为44.6%,对照组为42.9%,其中死亡发生率为1.1%和0.3%。帕博利珠单抗组中,3级及以上潜在免疫介导的不良事件发生率为10.0%。
结论:在局部晚期HNSCC患者中,加入新辅助和辅助帕博利珠单抗显著改善了事件无进展生存期。新辅助帕博利珠单抗不会影响手术完成的可能性。没有发现新的安全性信号。该研究由默克公司(Merck Sharp and Dohme)资助,KEYNOTE-689临床试验号为NCT03765918。
Safety and Efficacy of Obicetrapib in Patients at High Cardiovascular Risk
Obicetrapib在高心血管风险患者中的安全性和疗效
澳大利亚Monash大学维多利亚心脏研究所等机构联合发表
Abstract
Background: Obicetrapib is a highly selective cholesteryl ester transfer protein inhibitor that reduces low-density lipoprotein (LDL) cholesterol levels. The efficacy and safety of obicetrapib have not been fully characterized among patients at high risk for cardiovascular events.
Methods: We conducted a multinational, randomized, placebo-controlled trial involving patients with heterozygous familial hypercholesterolemia or a history of atherosclerotic cardiovascular disease who were receiving maximum tolerated doses of lipid-lowering therapy. Patients with an LDL cholesterol level of 100 mg per deciliter or higher or a non-high-density lipoprotein (HDL) cholesterol level of 130 mg per deciliter or higher, as well as those with an LDL cholesterol level of 55 to 100 mg per deciliter or a non-HDL cholesterol level of 85 to 130 mg per deciliter and at least one additional cardiovascular risk factor, were eligible for inclusion. The patients were randomly assigned in a 2:1 ratio to receive either 10 mg of obicetrapib once daily or matching placebo for 365 days. The primary end point was the percent change in the LDL cholesterol level from baseline to day 84.
Results: A total of 2530 patients underwent randomization; 1686 patients were assigned to receive obicetrapib and 844 to receive placebo. The mean age of the patients was 65 years, 34% were women, and the mean baseline LDL cholesterol level was 98 mg per deciliter. The least-squares mean percent change from baseline to day 84 in the LDL cholesterol level was -29.9% (95% confidence interval [CI], -32.1 to -27.8) in the obicetrapib group, as compared with 2.7% (95% CI, -0.4 to 5.8) in the placebo group, for a between-group difference of -32.6 percentage points (95% CI, -35.8 to -29.5; P<0.001). The incidence of adverse events appeared to be similar in the two groups.
Conclusions: Among patients with atherosclerotic cardiovascular disease or heterozygous familial hypercholesterolemia who were receiving maximum tolerated doses of lipid-lowering therapy and were at high risk for cardiovascular events, obicetrapib reduced LDL cholesterol levels by 29.9%. (Funded by NewAmsterdam Pharma; BROADWAY ClinicalTrials.gov number, NCT05142722.)
摘要
背景:Obicetrapib是一种高度选择性的胆固醇酯转移蛋白(CETP)抑制剂,可降低低密度脂蛋白(LDL)胆固醇水平。Obicetrapib在高心血管风险患者中的疗效和安全性尚未完全明确。
方法:我们进行了一项多国、随机、安慰剂对照试验,招募了患有杂合子家族性高胆固醇血症或动脉粥样硬化性心血管疾病病史,并接受最大耐受剂量降脂治疗的患者。符合入选标准的患者包括:低密度脂蛋白胆固醇(LDL-C)水平达到100毫克/分升或更高,或非高密度脂蛋白胆固醇(non-HDL-C)水平达到130毫克/分升或更高的患者,以及LDL-C水平在55至100毫克/分升之间,或non-HDL-C水平在85至130毫克/分升之间,且至少有一个额外心血管危险因素的患者。患者以2:1的比例随机分配,每日接受10毫克奥比曲泊(obicetrapib)或匹配的安慰剂,持续365天。主要终点是基线至第84天LDL-C水平的百分比变化。
结果:本研究共纳入2530例随机化患者,其中1686例分配至奥西卓泮治疗组,844例分配至安慰剂组。患者的平均年龄为65岁,其中34%为女性,基线LDL胆固醇水平的平均值为98 mg/dL。obicetrapib组从基线到第84天的LDL胆固醇水平的最小二乘均值变化百分比为-29.9%(95%置信区间[CI],-32.1至-27.8),而安慰剂组为2.7%(95% CI,-0.4至5.8),两组之间的差异为-32.6个百分点(95% CI,-35.8至-29.5;P<0.001)。两组的不良事件发生率相似。
结论:在接受最大耐受剂量脂质降低治疗并且处于高心血管风险的动脉粥样硬化性心血管疾病或异质性家族性高胆固醇血症患者中,obicetrapib将LDL胆固醇水平降低了29.9%。(资助方:NewAmsterdam Pharma;BROADWAY临床试验编号:NCT05142722。)
第393卷第2期一共发表31篇内容,其中原始研究4篇。
As-Needed Albuterol-Budesonide in Mild Asthma
按需使用阿布特罗-布地奈德用于治疗轻度哮喘

北卡罗来纳临床研究中心等机构联合发表
Abstract
BACKGROUND: As-needed use of albuterol-budesonide has been shown to result in a significantly lower risk of severe asthma exacerbation than as-needed use of albuterol alone among patients with moderate-to-severe asthma. Data on albuterol-budesonide in mild asthma are needed.
METHODS: We conducted a fully virtual, decentralized, phase 3b, multicenter, double-blind, event-driven trial involving persons 12 years of age or older with disease that was uncontrolled despite treatment for mild asthma with a short-acting [beta]2-agonist (SABA) with or without a low-dose inhaled glucocorticoid or leukotriene-receptor antagonist. Participants were randomly assigned in a 1:1 ratio to a fixed-dose combination of 180 [mu]g of albuterol and 160 [mu]g of budesonide (with each dose consisting of two inhaler actuations of 90 [mu]g and 80 [mu]g, respectively) or 180 [mu]g of albuterol (with each dose consisting of two inhaler actuations of 90 [mu]g) on an as-needed basis for up to 52 weeks. The primary end point was the first severe asthma exacerbation, assessed in a time-to-event analysis, in the on-treatment efficacy population, and the key secondary end point was the first severe exacerbation in the intention-to-treat population. Secondary end points included the annualized rate of severe asthma exacerbations and exposure to systemic glucocorticoids.
RESULTS: A total of 2516 participants underwent randomization; 1797 (71.4%) completed the trial. Of 2421 participants in the full analysis population (1209 assigned to the albuterol-budesonide group and 1212 to the albuterol group), 97.2% were 18 years of age or older; 74.4% used a SABA alone at baseline. The trial was stopped for efficacy at a prespecified interim analysis. A severe exacerbation occurred in 5.1% of the participants in the albuterol-budesonide group and in 9.1% of those in the albuterol group in the on-treatment efficacy population (hazard ratio, 0.53; 95% confidence interval [CI], 0.39 to 0.73) and in 5.3% and 9.4%, respectively, in the intention-to-treat population (hazard ratio, 0.54; 95% CI, 0.40 to 0.73) (P<0.001 for both comparisons). The annualized rate of severe asthma exacerbations was lower with albuterol-budesonide than with albuterol (0.15 vs. 0.32; rate ratio, 0.47; 95% CI, 0.34 to 0.64), as was the mean annualized total dose of systemic glucocorticoids (23.2 vs. 61.9 mg per year). Adverse events were similar in the two treatment groups.
CONCLUSIONS: As-needed use of albuterol-budesonide resulted in a lower risk of a severe asthma exacerbation than as-needed use of albuterol alone among participants with disease that was uncontrolled despite treatment for mild asthma. (Funded by Bond Avillion 2 Development and AstraZeneca; BATURA ClinicalTrials.gov number,NCT05505734)
摘要
背景:研究表明,按需使用沙丁胺醇-布地奈德组合药物比单独按需使用沙丁胺醇能够显著降低中度至重度哮喘患者的严重哮喘急性发作风险。然而,关于沙丁胺醇-布地奈德在轻度哮喘中的应用数据仍然不足。
方法:我们进行了一项完全虚拟、去中心化的III期b期多中心双盲事件驱动试验,参与者为12岁及以上、哮喘症状无法得到控制的患者,尽管他们已经接受了短效β2激动剂(SABA)治疗,且可能联合低剂量吸入性糖皮质激素或白三烯受体拮抗剂。参与者以1:1的比例随机分配到使用180μg沙丁胺醇和160μg布地奈德的固定剂量组合药物(每剂由90μg沙丁胺醇和80μg布地奈德组成)或180μg沙丁胺醇(每剂由90μg沙丁胺醇组成),按需使用,最多持续52周。主要终点是首次严重哮喘急性发作,采用时间事件分析法评估治疗中的有效性人群,关键次要终点是首次严重急性发作,评估意向治疗人群。次要终点包括年化严重哮喘急性发作率和系统性糖皮质激素的暴露。
结果:共有2516名参与者进行了随机分配;1797名(71.4%)完成了试验。在2421名完整分析人群的参与者中(1209名分配到沙丁胺醇-布地奈德组,1212名分配到沙丁胺醇组),97.2%为18岁及以上;74.4%的参与者在基线时仅使用SABA。试验在预定的中期分析时因显示疗效而提前停止。沙丁胺醇-布地奈德组的5.1%参与者发生了严重急性发作,而沙丁胺醇组为9.1%(风险比为0.53,95%置信区间为0.39至0.73),在意向治疗人群中,沙丁胺醇-布地奈德组为5.3%,沙丁胺醇组为9.4%(风险比为0.54,95%置信区间为0.40至0.73)(两组比较P<0.001)。沙丁胺醇-布地奈德组的年化严重哮喘急性发作率低于沙丁胺醇组(0.15 vs. 0.32;率比为0.47,95%置信区间为0.34至0.64),系统性糖皮质激素的年均总剂量也较低(23.2毫克 vs. 61.9毫克)。两组的不良事件相似。
结论:在治疗控制不佳的轻度哮喘患者中,按需使用沙丁胺醇-布地奈德比仅按需使用沙丁胺醇能够显著降低严重哮喘急性发作的风险。(由Bond Avillion 2 Development和AstraZeneca资助;BATURA临床试验编号NCT05505734)
Global Effect of Cardiovascular Risk Factors on Lifetime Estimates
心血管危险因素对预期寿命估计的全局性影响

全球心血管风险联盟联合发表
Abstract
BACKGROUND: Five risk factors account for approximately 50% of the global burden of cardiovascular disease. How the presence or absence of classic risk factors affects lifetime estimates of cardiovascular disease and death from any cause remains unclear.
METHODS: We harmonized individual-level data from 2,078,948 participants across 133 cohorts, 39 countries, and 6 continents. Lifetime risk of cardiovascular disease and death from any cause was estimated up to 90 years of age according to the presence or absence of arterial hypertension, hyperlipidemia, underweight and overweight or obesity, diabetes, and smoking at 50 years of age. Differences in life span (in terms of additional life-years free of cardiovascular disease or death from any cause) according to the presence or absence of these risk factors were also estimated. Risk-factor trajectories were analyzed to predict lifetime differences according to risk-factor variation.
RESULTS: The lifetime risk of cardiovascular disease was 24% (95% confidence interval [CI], 21 to 30) among women and 38% (95% CI, 30 to 45) among men for whom all five risk factors were present. In the comparison between participants with none of the risk factors and those with all the risk factors, the estimated number of additional life-years free of cardiovascular disease was 13.3 (95% CI, 11.2 to 15.7) for women and 10.6 (95% CI, 9.2 to 12.9) for men; the estimated number of additional life-years free of death was 14.5 (95% CI, 9.1 to 15.3) for women and 11.8 (95% CI, 10.1 to 13.6) for men. As compared with no changes in the presence of all risk factors, modification of hypertension at an age of 55 to less than 60 years was associated with the most additional life-years free of cardiovascular disease, and modification of smoking at an age of 55 to less than 60 years was associated with the most additional life-years free of death.
CONCLUSIONS: The absence of five classic risk factors at 50 years of age was associated with more than a decade greater life expectancy than the presence of all five risk factors, in both sexes. Persons who modified hypertension and smoking in midlife had the most additional life-years free of cardiovascular disease and death from any cause, respectively. (Funded by the German Center for Cardiovascular Research [DZHK]; ClinicalTrials.gov number,NCT05466825.)
摘要
背景:全球约50%的心血管疾病负担可归因于五个关键风险因素。然而,经典风险因素的存在与否如何影响个体终身心血管疾病及全因死亡的风险评估,目前仍未明确。
方法:我们整合了来自133个队列、39个国家和6大洲的2,078,948名参与者的个体数据。根据50岁时是否存在动脉高血压、高血脂、体重过轻或肥胖、糖尿病和吸烟,估算了心血管疾病和因任何原因死亡的终身风险,直到90岁。还估算了这些危险因素的存在或缺失对寿命的影响(以额外的无心血管疾病或无死亡年数为单位)。分析了危险因素的变化轨迹,以预测不同危险因素变动对寿命的影响。
结果:在所有五项危险因素均存在的女性中,心血管疾病的终身风险为24%(95%置信区间 [CI],21% 至 30%),男性为38%(95% CI,30% 至 45%)。在没有任何危险因素者与拥有所有危险因素者之间的比较中,估计女性额外无心血管疾病的生存年数为13.3年(95% CI,11.2 至 15.7),男性为10.6年(95% CI,9.2 至 12.9);估计女性额外无死亡的生存年数为14.5年(95% CI,9.1 至 15.3),男性为11.8年(95% CI,10.1 至 13.6)。相较于在所有危险因素均存在的情况下不做任何改变,55岁到不到60岁之间对高血压进行干预,与获得最多的额外无心血管疾病生存年数相关;55岁到不到60岁之间对吸烟进行干预,则与获得最多的额外无死亡生存年数相关。
结论:在50岁时不存在五种经典风险因素的人群中,无论男女,其预期寿命比存在所有五种风险因素的人群多出十余年。中年期间对高血压和吸烟进行干预的人群,分别获得了最多的无心血管疾病额外生存年数以及无任何原因死亡额外生存年数。(研究由德国心血管研究中心[DZHK]资助;临床试验注册号:NCT05466825)
Intravenous Tenecteplase before Thrombectomy in Stroke
溶栓前静脉注射替奈普酶联合血管内取栓治疗急性缺血性脑卒中的效果
中国陆军军医大学新桥医院神经内科等机构联合发表
Abstract
BACKGROUND: The safety and efficacy of treatment with intravenous tenecteplase before endovascular thrombectomy in patients with acute ischemic stroke due to large-vessel occlusion remain uncertain.
METHODS: In this open-label trial conducted in China, we randomly assigned patients with acute ischemic stroke due to large-vessel occlusion who had presented within 4.5 hours after onset and were eligible for thrombolysis to receive either intravenous tenecteplase followed by endovascular thrombectomy or endovascular thrombectomy alone. The primary outcome was functional independence (a score of 0 to 2 on the modified Rankin scale; range, 0 to 6, with higher scores indicating more severe disability) at 90 days. Secondary outcomes included successful reperfusion before and after thrombectomy. Safety outcomes included symptomatic intracranial hemorrhage within 48 hours and death within 90 days.
RESULTS: A total of 278 patients were randomly assigned to the tenecteplase-thrombectomy group and 272 to the thrombectomy-alone group. Functional independence at 90 days was observed in 147 patients (52.9%) in the tenecteplase-thrombectomy group and in 120 patients (44.1%) in the thrombectomy-alone group (unadjusted risk ratio, 1.20; 95% confidence interval, 1.01 to 1.43; P=0.04). A total of 6.1% of the patients in the tenecteplase-thrombectomy group and 1.1% of those in the thrombectomy-alone group had successful reperfusion before thrombectomy, and 91.4% and 94.1%, respectively, had successful reperfusion after thrombectomy. Symptomatic intracranial hemorrhage within 48 hours occurred in 8.5% of the patients in the tenecteplase-thrombectomy group and in 6.7% of those in the thrombectomy-alone group; mortality at 90 days was 22.3% and 19.9%, respectively.
CONCLUSIONS: Among patients with acute ischemic stroke due to large-vessel occlusion who had presented within 4.5 hours after onset, the percentage of patients with functional independence at 90 days was higher with intravenous tenecteplase plus endovascular thrombectomy than with endovascular thrombectomy alone. (Funded by the Chongqing Science and Health Joint Medical Research Project and others; BRIDGE-TNK ClinicalTrials.gov number,NCT04733742.)
摘要
背景: 在因大血管闭塞引起的急性缺血性脑卒中患者中,静脉注射替奈普酶(tenecteplase)联合血管内取栓术的安全性和疗效仍不确定。
方法: 本研究是一项在中国开展的开放标签临床试验。我们将符合溶栓治疗标准且在发病后4.5小时内就诊的急性缺血性脑卒中患者,随机分配至两组:一组接受静脉注射替奈普酶后进行血管内取栓术(替奈普酶+血管内取栓组),另一组仅接受血管内取栓术(血管内取栓组)。主要结局是90天时的功能独立性(改良Rankin量表评分0到2,评分范围0到6,分数越高代表残疾越严重)。次要结局包括血管内取栓术前后成功再灌注的情况。安全性结局包括在48小时内出现症状性颅内出血和90天内死亡的情况。
结果: 共有278例患者被随机分配至替奈普酶联合取栓组,272例分配至单纯取栓组。替奈普酶联合取栓组中有147例患者(52.9%)、单纯取栓组中有120例患者(44.1%)在90天时达到功能独立(未调整风险比1.20;95%置信区间1.01至1.43;P=0.04)。替奈普酶联合取栓组中有6.1%的患者在取栓术前实现成功再灌注,单纯取栓组为1.1%;取栓术后两组成功率分别为91.4%和94.1%。替奈普酶联合取栓组中8.5%的患者在48小时内出现症状性颅内出血,单纯取栓组为6.7%;90天死亡率分别为22.3%和19.9%。
结论: 在因大血管闭塞引起的急性缺血性脑卒中患者中,若患者在发病后4.5小时内到达医院,接受静脉注射替奈普酶联合血管内取栓术的患者比单纯接受血管内取栓术的患者,90天时功能独立性更高。(研究由重庆市科技卫生联合医学研究项目等资助;BRIDGE-TNK 临床试验号,NCT04733742。)
Overall Survival with Inavolisib in PIK3CA-Mutated Advanced Breast CancerInavolisib 治疗 PIK3CA 突变晚期乳腺癌的总生存期数据
美国纪念斯隆凯特琳癌症中心等机构联合发表
Abstract
BACKGROUND: In the phase 3, double-blind, randomized INAVO120 trial, treatment with inavolisib plus palbociclib-fulvestrant led to a significant progression-free survival benefit, as compared with placebo plus palbociclib-fulvestrant, among patients with PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer who had had relapse during or within 12 months after completion of adjuvant endocrine therapy.
METHODS: We randomly assigned patients with PIK3CA-mutated, hormone receptor-positive, HER2-negative locally advanced or metastatic breast cancer who had had disease recurrence or progression during or within 12 months after completion of adjuvant endocrine therapy to receive inavolisib plus palbociclib-fulvestrant (inavolisib group) or placebo plus palbociclib-fulvestrant (placebo group). In the current report, we provide the results of the final analysis of overall survival, including updated data on efficacy and safety.
RESULTS: A total of 161 patients were assigned to the inavolisib group, and 164 to the placebo group. After a median follow-up of 34.2 months in the inavolisib group and 32.3 months in the placebo group, the median overall survival was 34.0 months (95% confidence interval [CI], 28.4 to 44.8) with inavolisib and 27.0 months (95% CI, 22.8 to 38.7) with placebo (hazard ratio for death, 0.67; 95% CI, 0.48 to 0.94; P=0.02 [prespecified boundary for statistical significance, P<0.0469]). An objective response occurred in 62.7% (95% CI, 54.8 to 70.2) of patients in the inavolisib group and 28.0% (95% CI, 21.3 to 35.6) of those in the placebo group (P<0.001). The updated hazard ratio for disease progression or death was 0.42 (95% CI, 0.32 to 0.55). Adverse events led to discontinuation of inavolisib in 6.8% of patients and discontinuation of placebo in 0.6%. The incidence of hyperglycemia, stomatitis or mucosal inflammation, gastrointestinal toxic effects (e.g., diarrhea), and ocular toxic effects (e.g., dry eye and blurred vision) was higher with inavolisib than with placebo.
CONCLUSIONS: Treatment with inavolisib plus palbociclib-fulvestrant led to a significant overall survival benefit, as compared with placebo plus palbociclib-fulvestrant. Hyperglycemia, stomatitis or mucosal inflammation, gastrointestinal toxic effects, and ocular toxic effects were reported more frequently with inavolisib than with placebo. (Funded by F. Hoffmann-La Roche; INAVO120 ClinicalTrials.gov number,NCT04191499.)
摘要
背景:在III期双盲随机临床试验INAVO120中,针对PIK3CA突变、激素受体阳性、人表皮生长因子受体2(HER2)阴性的局部晚期或转移性乳腺癌患者(既往在辅助内分泌治疗期间或完成治疗后12个月内复发),与安慰剂联合帕博西利-氟维司群相比,inavolisib联合帕博西利-氟维司群治疗显著延长了无进展生存期。
方法:我们将PIK3CA突变、激素受体阳性、HER2阴性局部晚期或转移性乳腺癌患者(这些患者在或在完成辅助内分泌治疗后12个月内出现疾病复发或进展)随机分配到接受Inavolisib联合帕博昔布-氟维司群(Inavolisib组)或安慰剂联合帕博昔布-氟维司群(安慰剂组)。在本报告中,我们提供了总体生存期的最终分析结果,包括更新的疗效和安全性数据。
结果:共161名患者被分配到Inavolisib组,164名患者被分配到安慰剂组。经过34.2个月(Inavolisib组)和32.3个月(安慰剂组)的中位随访后,Inavolisib组的中位总体生存期为34.0个月(95%置信区间[CI],28.4至44.8个月),安慰剂组为27.0个月(95% CI,22.8至38.7个月)(死亡风险比为0.67;95% CI,0.48至0.94;P=0.02[统计学显著性预设边界为P<0.0469])。Inavolisib组62.7%的患者(95% CI,54.8至70.2)出现了客观反应,而安慰剂组为28.0%(95% CI,21.3至35.6)(P<0.001)。更新后的疾病进展或死亡的风险比为0.42(95% CI,0.32至0.55)。有6.8%的Inavolisib组患者和0.6%的安慰剂组患者因不良事件停药。Inavolisib组的高血糖、口腔炎或黏膜炎、胃肠道毒性(如腹泻)和眼部毒性(如干眼和视力模糊)发生率高于安慰剂组。
结论:Inavolisib联合帕博昔布-氟维司群治疗相比安慰剂联合帕博昔布-氟维司群治疗,显著改善了总体生存期。高血糖、口腔炎或黏膜炎、胃肠道毒性和眼部毒性在Inavolisib组的发生频率高于安慰剂组。(由F. Hoffmann-La Roche资助;INAVO120临床试验号,NCT04191499。)
第393卷第3期一共发表20篇内容,其中原始研究4篇。
Perioperative Durvalumab in Gastric and Gastroesophageal Junction Cancer
围手术期杜瓦利单抗在胃癌及胃食管交界处癌中的应用
美国纪念斯隆凯特琳癌症中心等机构联合发表
Abstract
BACKGROUND: Perioperative FLOT (fluorouracil, leucovorin, oxaliplatin, and docetaxel) is a standard therapy for resectable gastric and gastroesophageal junction adenocarcinomas, but recurrence rates remain high. Immunotherapy plus chemotherapy may improve outcomes.
METHODS: In a phase 3, multinational, double-blind, randomized trial, we assigned participants with resectable gastric or gastroesophageal junction adenocarcinoma, in a 1:1 ratio, to receive durvalumab at a dose of 1500 mg or placebo every 4 weeks plus FLOT for 4 cycles (2 cycles each of neoadjuvant and adjuvant therapy), followed by durvalumab or placebo every 4 weeks for 10 cycles. The primary end point was event-free survival; secondary end points included overall survival and pathological complete response.
RESULTS: A total of 474 participants were randomly assigned to the durvalumab group, and 474 to the placebo group (median follow-up, 31.5 months; interquartile range, 26.7 to 36.6). Two-year event-free survival (Kaplan-Meier estimate) was 67.4% among the participants in the durvalumab group and 58.5% among those in the placebo group (hazard ratio for event or death, 0.71; 95% confidence interval [CI], 0.58 to 0.86; P<0.001). Two-year overall survival was 75.7% in the durvalumab group and 70.4% in the placebo group (piecewise hazard ratio for death during months 0 to 12, 0.99 [95% CI, 0.70 to 1.39], and during the period from month 12 onward, 0.67 [95% CI, 0.50 to 0.90]; P=0.03 by a stratified log-rank test [exceeding the significance threshold of P<0.0001]). The percentage of participants with a pathological complete response was 19.2% in the durvalumab group and 7.2% in the placebo group (relative risk, 2.69 [95% CI, 1.86 to 3.90]). Adverse events with a maximum grade of 3 or 4 were reported in 340 participants (71.6%) in the durvalumab group and in 334 (71.2%) in the placebo group. The percentage of participants with delayed surgery was 10.1% and 10.8%, respectively, and the percentage with delayed initiation of adjuvant treatment was 2.3% and 4.6%.
CONCLUSIONS: Perioperative durvalumab plus FLOT led to significantly better event-free survival outcomes than FLOT alone among participants with resectable gastric or gastroesophageal junction adenocarcinoma. (Funded by AstraZeneca; MATTERHORN ClinicalTrials.gov number,NCT04592913.)
摘要
背景:围手术期FLOT方案(氟尿嘧啶、亚叶酸钙、奥沙利铂、多西他赛)是可切除胃癌和胃食管交界腺癌的标准治疗方法,但复发率仍然较高。免疫治疗联合化疗可能改善治疗结局。
方法:我们开展了一项多国、多中心、双盲、随机的III期临床试验,将可切除的胃癌或胃食管交界腺癌患者按1:1比例分组,接受Durvalumab(1500毫克,每4周一次)或安慰剂治疗,并联合FLOT方案治疗4个周期(术前和术后各2个周期),随后继续每4周一次使用Durvalumab或安慰剂共10个周期。主要终点为无事件生存期(event-free survival),次要终点包括总生存期和病理学完全缓解率。
结果:共948名受试者被随机分为Durvalumab组(474人)和安慰剂组(474人),中位随访时间为31.5个月(四分位范围26.7–36.6个月)。两年无事件生存率(Kaplan–Meier估计)为Durvalumab组67.4%,安慰剂组为58.5%(事件或死亡的风险比为0.71;95%置信区间[CI],0.58–0.86;P<0.001)。两年总生存率为Durvalumab组75.7%,安慰剂组为70.4%(分段风险比:0–12个月内为0.99 [95% CI, 0.70–1.39];12个月以后为0.67 [95% CI, 0.50–0.90];按分层log-rank检验P=0.03,高于预设显著性阈值P<0.0001)。病理学完全缓解率为Durvalumab组19.2%,安慰剂组为7.2%(相对风险为2.69;95% CI, 1.86–3.90)。3或4级不良事件的发生率分别为71.6%(Durvalumab组)和71.2%(安慰剂组)。手术延迟发生率分别为10.1%和10.8%;辅助治疗起始延迟发生率分别为2.3%和4.6%。
结论:在可切除的胃癌或胃食管交界腺癌患者中,围手术期使用Durvalumab联合FLOT治疗相比单纯FLOT治疗,能显著改善无事件生存期。(本研究由阿斯利康公司资助;临床试验注册号:NCT04592913)
First-Line Treatment of Pulmonary Sarcoidosis with Prednisone or Methotrexate
泼尼松或甲氨蝶呤作为肺结节病一线治疗的比较研究
鹿特丹Erasmus医学中心等机构联合发表
Abstract
BACKGROUND: Prednisone is currently recommended as the first-line treatment for pulmonary sarcoidosis but is associated with many side effects. Methotrexate, which is recommended as a second-line treatment, appears to have fewer side effects than prednisone but a slower onset of action. Data are needed on the efficacy and side-effect profile of methotrexate as compared with prednisone as first-line treatment for pulmonary sarcoidosis.
METHODS: In this multicenter, open-label, noninferiority trial involving patients with pulmonary sarcoidosis who had not previously received treatment, we randomly assigned patients, in a 1:1 ratio, to receive prednisone or methotrexate according to a prespecified treatment schedule. The primary end point was the mean change from baseline to week 24 in the percentage of the predicted forced vital capacity (FVC), as estimated with the use of mixed models for repeated measures. The noninferiority margin for the primary end point was 5 percentage points.
RESULTS: Of the 138 patients who underwent randomization, 70 were assigned to receive prednisone and 68 to receive methotrexate. The unadjusted mean change from baseline to week 24 in the percentage of the predicted FVC was 6.75 percentage points (95% confidence interval [CI], 4.50 to 8.99) in the prednisone group and 6.11 percentage points (95% CI, 3.72 to 8.50) in the methotrexate group. Methotrexate was noninferior to prednisone with regard to the primary end point, with an adjusted between-group difference of -1.17 percentage points (95% CI, -4.27 to 1.93). Adverse events occurred in a similar percentage of patients in the two trial groups. Weight gain, insomnia, and increased appetite were the most common adverse events with prednisone, and nausea, fatigue, and any abnormal liver-function test were among the most common adverse events with methotrexate.
CONCLUSIONS: In patients with pulmonary sarcoidosis, initial treatment with methotrexate was noninferior to that with prednisone with regard to the change from baseline to week 24 in the percentage of the predicted FVC. Differences in the side-effect profile between methotrexate and prednisone may inform shared decision making by providers and patients about the appropriate treatment approach. (Funded by the Dutch Lung Foundation; PREDMETH ClinicalTrials.gov number,NCT04314193.)
摘要
背景:目前推荐泼尼松(prednisone)作为肺结节病的一线治疗药物,但其副作用较多。甲氨蝶呤(methotrexate)作为二线治疗药物,副作用似乎少于泼尼松,但起效较慢。目前尚缺乏将甲氨蝶呤与泼尼松用于肺结节病一线治疗的疗效和副作用对比数据。
方法:这是一项多中心、开放标签的非劣效性试验,纳入了此前未接受治疗的肺结节病患者。患者按1:1比例随机分配至泼尼松组或甲氨蝶呤组,按照预设的治疗方案进行治疗。主要终点是第24周时预测用力肺活量(FVC)百分比相较基线的平均变化,采用重复测量混合模型估算。非劣效界值设为5个百分点。
结果:在138例接受随机分组的患者中,70例被分配接受泼尼松治疗,68例接受甲氨蝶呤治疗。泼尼松组预测用力肺活量(FVC)百分比从基线至第24周未经校正的平均变化为6.75个百分点(95%置信区间[CI],4.50至8.99),甲氨蝶呤组为6.11个百分点(95% CI,3.72至8.50)。就主要终点而言,甲氨蝶呤非劣效于泼尼松,校正后的组间差异为-1.17个百分点(95% CI,-4.27至1.93)。两组患者中发生不良事件的比例相近。泼尼松组最常见的不良事件为体重增加、失眠和食欲增强,而甲氨蝶呤组最常见的不良事件包括恶心、疲劳及肝功能检查异常。
结论:在肺结节病患者中,甲氨蝶呤用于初始治疗在改善第24周预测FVC百分比方面不劣于泼尼松。甲氨蝶呤和泼尼松在副作用谱方面存在差异,这些信息可为医生和患者在选择合适治疗方案时提供依据。(本研究由荷兰肺基金会资助;PREDMETH试验注册号:NCT04314193)
Graft-versus-Host Disease Prophylaxis with Cyclophosphamide and Cyclosporin
环磷酰胺联合环孢素用于移植物抗宿主病的预防
澳大利亚与新西兰白血病和淋巴瘤研究协作组等机构联合发表
Abstract
BACKGROUND: Allogeneic peripheral-blood stem-cell transplantation (SCT) from a matched related donor after myeloablative conditioning is the preferred curative treatment for patients with high-risk blood cancers. The combination of a calcineurin inhibitor and an antimetabolite remains standard care for graft-versus-host disease (GVHD) prophylaxis in these patients. Data from two randomized trials have suggested that post-transplantation cyclophosphamide can reduce the risk of GVHD after SCT from a matched donor when it is added to or replaces the antimetabolite. However, the effects of post-transplantation cyclophosphamide specifically after SCT from a matched related donor remain uncertain, and effects in the context of myeloablative conditioning are unclear.
METHODS: We randomly assigned adults who were undergoing SCT from a matched related donor after myeloablative or reduced-intensity conditioning to receive either post-transplantation cyclophosphamide-cyclosporin (experimental prophylaxis) or cyclosporin-methotrexate (standard prophylaxis). The primary end point was GVHD-free, relapse-free survival.
RESULTS: Among 134 patients who underwent randomization, 66 were assigned to receive experimental prophylaxis and 68 to receive standard prophylaxis. GVHD-free, relapse-free survival was significantly longer with experimental prophylaxis (median, 26.2 months; 95% confidence interval [CI], 9.1 to not reached) than with standard prophylaxis (median, 6.4 months; 95% CI, 5.6 to 8.3; P<0.001 by a log-rank test). GVHD-free, relapse-free survival at 3 years was 49% (95% CI, 36 to 61) with experimental prophylaxis and 14% (95% CI, 6 to 25) with standard prophylaxis (hazard ratio for GVHD, relapse, or death, 0.42; 95% CI, 0.27 to 0.66). The cumulative incidence of grade III to IV acute GVHD at 3 months was 3% (95% CI, 1 to 10) in the experimental-prophylaxis group and 10% (95% CI, 4 to 19) in the standard-prophylaxis group. At 2 years, overall survival was 83% and 71%, respectively (hazard ratio for death, 0.59; 95% CI, 0.29 to 1.19). The incidence of serious adverse events was similar in the two groups in the first 100 days after SCT.
CONCLUSIONS: The combination of post-transplantation cyclophosphamide and a calcineurin inhibitor led to longer GVHD-free, relapse-free survival than standard prophylaxis after transplantation from a matched related donor with either reduced-intensity or myeloablative conditioning in patients with blood cancers. (Funded by the Australian Government Medical Research Future Fund and others; ALLG BM12 CAST Australian-New Zealand Clinical Trials Registry number,ACTRN12618000505202.)
摘要
背景:来自匹配的近亲供者的异基因外周血干细胞移植(SCT),在进行预处理性骨髓消融性清除(myeloablative conditioning)后,是高危血癌患者的首选根治性治疗方法。钙调磷酸酶抑制剂联合抗代谢药物的联合治疗,在这些患者中仍然是移植物抗宿主病(GVHD)预防的标准疗法。来自两项随机试验的数据表明,移植后环磷酰胺(post-transplantation cyclophosphamide)在与抗代谢药物联合使用或替代抗代谢药物时,可以降低来自匹配供者的SCT后GVHD的风险。然而,移植后环磷酰胺在特别来自匹配的近亲供者的SCT中的效果仍不确定,并且在预处理性骨髓消融性清除的背景下的效果也不清楚。
方法:研究将接受亲属相合供者移植并经过强效或减毒清髓预处理的成年患者,随机分配接受两种GVHD预防方案之一:实验组使用移植后环磷酰胺联合环孢素(PTCy–环孢素),对照组使用标准方案环孢素联合甲氨蝶呤。主要终点为“无GVHD、无复发的生存期”(GVHD-free, relapse-free survival, GRFS)。
结果:共134名患者接受随机分组,其中66人接受实验性方案,68人接受标准方案。实验组的GRFS显著延长(中位数为26.2个月;95%置信区间[CI]:9.1–未达到),而标准组为6.4个月(95% CI:5.6–8.3;P<0.001)。三年时,实验组的GRFS为49%(95% CI:36–61),而标准组为14%(95% CI:6–25),GVHD、复发或死亡的风险比为0.42(95% CI:0.27–0.66)。三个月内III–IV级急性GVHD的累计发生率分别为实验组3%(95% CI:1–10)和标准组10%(95% CI:4–19)。两年总生存率为实验组83%,标准组71%(死亡风险比0.59;95% CI:0.29–1.19)。移植后前100天内,两组严重不良事件发生率相似。
结论:在患有血液癌症、接受亲属相合供者移植的患者中,无论采用减毒或强效清髓预处理,移植后使用环磷酰胺联合钙调神经磷酸酶抑制剂可显著延长无GVHD、无复发的生存时间,优于传统的GVHD预防方案。(本研究由澳大利亚政府医学研究未来基金等资助;试验注册号ACTRN12618000505202)
Multidose Ondansetron after Emergency Visits in Children with Gastroenteritis儿童胃肠炎急诊后多剂量昂丹司琼的使用
加拿大 Pediatric Emergency Research Canada(PERC)研究网络发起并组织
Abstract
BACKGROUND: Ondansetron improves outcomes when administered in emergency departments to children with acute gastroenteritis-associated vomiting. It is commonly prescribed at discharge to reduce symptoms, but evidence to support this practice is limited.
METHODS: We conducted a double-blind, randomized superiority trial involving children 6 months to less than 18 years of age with acute gastroenteritis-associated vomiting in six pediatric emergency departments. Caregivers were provided with six doses of oral ondansetron or placebo to administer in response to ongoing vomiting during the first 48 hours after enrollment. The primary outcome was moderate-to-severe gastroenteritis, defined by a score of 9 or higher on the modified Vesikari scale (scores range from 0 to 20, with higher scores indicating greater severity), during the 7 days after enrollment. Secondary outcomes included the presence of vomiting, the duration of vomiting (defined as the time from enrollment to the last vomiting episode), the number of vomiting episodes within 48 hours after enrollment, unscheduled physician visits within 7 days after enrollment, and receipt of intravenous fluids.
RESULTS: A total of 1030 children underwent randomization. Moderate-to-severe gastroenteritis occurred in 5.1% (23 of 452 participants for whom data were available) in the ondansetron group and 12.5% (55 of 441) in the placebo group (unadjusted risk difference, -7.4 percentage points; 95% confidence interval [CI], -11.2 to -3.7). After adjustment for site, weight, and missing data, ondansetron was associated with a lower risk of moderate-to-severe gastroenteritis than placebo (adjusted odds ratio, 0.50; 95% CI, 0.40 to 0.60). Although we did not observe any meaningful difference between the groups in the presence or median duration of vomiting, the total number of vomiting episodes within 48 hours after enrollment was lower with ondansetron than with placebo (adjusted rate ratio, 0.76; 95% CI, 0.67 to 0.87). The percentage of children who had unscheduled health care visits and the percentage who received intravenous fluids after enrollment did not differ substantially between the groups. The incidence of adverse events also did not differ meaningfully between the groups (odds ratio, 0.99; 95% CI, 0.61 to 1.61).
CONCLUSIONS: Among children with gastroenteritis-associated vomiting, the provision of ondansetron after an emergency department visit led to lower risk of moderate-to-severe gastroenteritis during the subsequent 7 days than the provision of placebo. (Funded by the Canadian Institutes of Health Research and others; ClinicalTrials.gov number,NCT03851835.)
摘要
背景:在急诊科,昂丹司琼(Ondansetron)可改善患有急性胃肠炎相关呕吐的儿童的治疗结果。为减少症状,该药常在出院时处方,但支持这种做法的证据有限。
方法:本研究是一项双盲、随机、优效性试验,纳入6个月至18岁以下、因急性胃肠炎相关呕吐而就诊的儿童,地点为六家儿科急诊中心。研究向患儿看护者提供6剂口服昂丹司琼或安慰剂,供在入组后48小时内持续呕吐时按需服用。主要结局为入组后7天内出现中度至重度胃肠炎(经修订的Vesikari评分≥9,评分范围为0–20,分数越高表示病情越重)。次要结局包括是否出现呕吐、呕吐持续时间(从入组至最后一次呕吐的时间)、入组后48小时内呕吐的次数、7天内是否有非计划的就医、是否接受静脉补液治疗。
结果:共有1030名儿童被随机分组。在有完整数据的参与者中,昂丹司琼组中有5.1%(23/452人)发生中度至重度胃肠炎,而安慰剂组为12.5%(55/441人),未调整的风险差为−7.4个百分点(95%置信区间,−11.2 至 −3.7)。经地点、体重和缺失数据调整后,昂丹司琼与较低的中重度胃肠炎风险相关(调整后比值比为0.50;95%置信区间,0.40–0.60)。虽然两组在呕吐发生率和呕吐中位持续时间方面无明显差异,但入组后48小时内的呕吐总次数在昂丹司琼组明显少于安慰剂组(调整后的比率比为0.76;95%置信区间,0.67–0.87)。两组在非计划就医率和静脉补液率方面无显著差异。不良事件发生率在两组间也无明显差别(比值比为0.99;95%置信区间,0.61–1.61)。
结论:对于因胃肠炎引起呕吐的儿童,在急诊就诊后提供昂丹司琼治疗,在随后的7天内可降低发生中度至重度胃肠炎的风险,相较于给予安慰剂更为有效。(本研究由加拿大卫生研究院等资助;ClinicalTrials.gov注册号:NCT03851835)
第393卷第4期一共发表27篇内容,其中原始研究4篇。
Weekly Fixed-Dose Insulin Efsitora in Type 2 Diabetes without Previous Insulin Therapy
无既往胰岛素治疗的2型糖尿病患者中每周固定剂量胰岛素 Efsitora 的研究
美国德克萨斯州达拉斯的Velocity临床研究机构等机构联合发表
Abstract
Background: In previous treat-to-target trials, adjustments to the dose of basal insulin have been made at least weekly, according to fasting blood glucose levels. A fixed-dose regimen of insulin efsitora alfa (efsitora), a once-weekly basal insulin, may provide a benefit in adults with type 2 diabetes who have not received previous insulin therapy.
Methods: We conducted a 52-week, phase 3, open-label, treat-to-target trial involving adults with type 2 diabetes who had not previously received insulin therapy. Participants were randomly assigned in a 1:1 ratio to receive once-weekly efsitora or once-daily insulin glargine U100 (glargine). Treatment with efsitora was initiated as a single dose of 100 U administered once weekly, with dose adjustments made every 4 weeks, as needed, at fixed doses of 150, 250, and 400 U to achieve fasting blood glucose levels of 80 to 130 mg per deciliter. Doses of glargine were adjusted weekly or more often according to a standard algorithm to reach the same glycemic goals. The primary end point, tested for noninferiority (noninferiority margin, 0.4 percentage points), was the change from baseline in the glycated hemoglobin level at 52 weeks.
Results: A total of 795 participants underwent randomization. The mean glycated hemoglobin level decreased from 8.20% at baseline to 7.05% at week 52 with efsitora (least-squares mean change, -1.19 percentage points) and from 8.28% to 7.08% with glargine (least-squares mean change, -1.16 percentage points); the estimated between-group difference of -0.03 percentage points (95% confidence interval [CI], -0.18 to 0.12) confirmed the noninferiority of efsitora to glargine. Superiority was not shown (P=0.68). The rate of combined clinically significant hypoglycemia (glucose level,<54 mg per deciliter) or severe hypoglycemia (level 3; requiring assistance for treatment) was lower with efsitora than with glargine (0.50 events per participant-year of exposure with efsitora vs. 0.88 with glargine; estimated rate ratio, 0.57 [95% CI, 0.39 to 0.84]). At week 52, the mean total weekly insulin dose was 289.1 U per week with efsitora and 332.8 U per week with glargine (estimated between-group difference, -43.7 U per week; 95% CI, -62.4 to -25.0); the median number of dose adjustments needed was 2 with efsitora and 8 with glargine.
Conclusions: In adults with type 2 diabetes who had not previously received insulin, once-weekly efsitora, administered in a fixed-dose regimen, was noninferior to once-daily glargine in reducing glycated hemoglobin levels. (Funded by Eli Lilly; ClinicalTrials.gov number, NCT05662332.)
摘要
背景:在以目标为导向的既往研究中,基础胰岛素的剂量调整通常依据空腹血糖水平至少每周进行一次。每周一次给药的基础胰岛素 efsitora alfa(efsitora)若采用固定剂量方案,可能为尚未接受过胰岛素治疗的2型糖尿病成人患者带来益处。
方法:本研究为一项为期52周的、开放标签、以治疗目标为导向的3期临床试验,纳入了未曾接受胰岛素治疗的2型糖尿病成人患者。参与者按1:1比例随机分配至每周一次 efsitora 或每日一次甘精胰岛素U100(glargine)治疗组。efsitora 起始剂量为每周100单位,随后每4周根据需要进行剂量调整,固定调整至150、250 或 400单位,以实现空腹血糖控制在80–130 mg/dL范围内。glargine 的剂量则依据标准算法每周或更频繁地进行调整,目标为相同的血糖控制范围。主要终点为52周时糖化血红蛋白(HbA1c)相较基线的变化,并以非劣效性(非劣效界值为0.4个百分点)为检验标准。
结果:共有795名受试者完成随机分组。efsitora 组的平均 HbA1c 水平从基线的8.20%下降至52周时的7.05%(最小二乘均值变化为−1.19个百分点),glargine 组则从8.28%下降至7.08%(最小二乘均值变化为−1.16个百分点);两组之间的估计差值为−0.03个百分点(95%置信区间,−0.18 至 0.12),证实了 efsitora 相对于 glargine 的非劣效性,未显示出优效性(P=0.68)。临床显著低血糖(血糖<54 mg/dL)或严重低血糖(3级;需他人协助治疗)事件发生率在 efsitora 组低于 glargine 组(每位参与者每年暴露事件数:0.50 vs. 0.88;估计事件比率为 0.57,95%置信区间,0.39 至 0.84)。在第52周时,efsitora 组的平均总每周胰岛素用量为289.1单位,glargine 组为332.8单位(两组间估计差值为−43.7单位,95%置信区间,−62.4 至 −25.0);所需的剂量调整中位数为 efsitora 组2次,glargine 组8次。
结论:对于未曾接受胰岛素治疗的2型糖尿病成人患者,每周一次、固定剂量给药的 efsitora 在降低 HbA1c 水平方面,与每日一次的 glargine 相比具有非劣效性。(本研究由礼来公司资助;ClinicalTrials.gov 登记号:NCT05662332)
Trastuzumab Deruxtecan or Ramucirumab plus Paclitaxel in Gastric Cancer
曲妥珠单抗德鲁司他肯或雷莫芦单抗联合紫杉醇用于胃癌治疗
日本国立癌症中心东医院等机构联合发表
Abstract
Background: On the basis of phase 2 studies, trastuzumab deruxtecan was approved for patients with human epidermal growth factor receptor 2 (HER2)-positive metastatic gastric cancer or gastroesophageal junction adenocarcinoma who had previously received trastuzumab-based therapy. Ramucirumab plus paclitaxel is also a standard second-line treatment option regardless of HER2 status.
Methods: We conducted an international, randomized, phase 3 trial comparing second-line trastuzumab deruxtecan at a dose of 6.4 mg per kilogram of body weight with ramucirumab plus paclitaxel in patients with HER2-positive metastatic gastric cancer or gastroesophageal junction adenocarcinoma confirmed on tumor biopsy conducted after the patient had progression while receiving trastuzumab-based therapy. The primary end point was overall survival. Secondary end points included progression-free survival, confirmed objective response (complete or partial response lasting >=4 weeks), disease control, duration of response, and safety.
Results: Among 494 patients who had undergone randomization, overall survival was significantly longer with trastuzumab deruxtecan than with ramucirumab plus paclitaxel (median, 14.7 vs. 11.4 months; hazard ratio for death, 0.70; 95% confidence interval, 0.55 to 0.90; P=0.004). Significant results were also seen with regard to progression-free survival (hazard ratio for disease progression or death, 0.74; 95% CI, 0.59 to 0.92) and confirmed objective response (in 44.3% of the patients in the trastuzumab deruxtecan group vs. 29.1% of those in the ramucirumab-paclitaxel group). The incidence of drug-related adverse events of any grade was 93.0% with trastuzumab deruxtecan and 91.4% with ramucirumab plus paclitaxel; the incidence of drug-related adverse events of grade 3 or higher was 50.0% and 54.1%, respectively. Adjudicated drug-related interstitial lung disease or pneumonitis occurred in 13.9% of the patients who received trastuzumab deruxtecan (grade 1 or 2 in 33 patients and grade 3 in 1) and in 1.3% of those who received ramucirumab plus paclitaxel (grade 3 in 2 patients and grade 5 in 1).
Conclusions: Trastuzumab deruxtecan led to significantly longer overall survival than ramucirumab plus paclitaxel among patients with HER2-positive metastatic gastric cancer or gastroesophageal junction adenocarcinoma. Adverse events were common in both groups. Events of interstitial lung disease or pneumonitis with trastuzumab deruxtecan, a known risk, were mainly low-grade. (Funded by Daiichi Sankyo and AstraZeneca; DESTINY-Gastric04 ClinicalTrials.gov number, NCT04704934.)
摘要
背景:基于既往的Ⅱ期研究结果,曲妥珠单抗德鲁司他肯(trastuzumab deruxtecan)已获批准用于既往接受过曲妥珠单抗方案治疗的人类表皮生长因子受体2(HER2)阳性转移性胃癌或胃食管结合部腺癌患者。雷莫芦单抗联合紫杉醇(ramucirumab plus paclitaxel)亦为标准的二线治疗方案,适用于无论HER2状态如何的患者。
方法:我们开展了一项国际、多中心、随机的Ⅲ期临床试验,比较二线治疗中曲妥珠单抗德鲁司他肯(剂量为6.4 mg/kg)与雷莫芦单抗联合紫杉醇的疗效。入组对象为经肿瘤活检确认为HER2阳性,且在接受曲妥珠单抗治疗后出现疾病进展的转移性胃癌或胃食管结合部腺癌患者。主要终点为总生存期(overall survival, OS);次要终点包括无进展生存期(progression-free survival, PFS)、确认的客观缓解率(完全缓解或部分缓解,持续≥4周)、疾病控制率、缓解持续时间及安全性等。
结果:共494例患者完成随机分组。与雷莫芦单抗联合紫杉醇相比,曲妥珠单抗德鲁司他肯治疗组的中位总生存期显著延长(14.7个月 vs. 11.4个月;死亡风险比 [HR] = 0.70;95%置信区间 [CI]:0.55–0.90;P = 0.004)。在无进展生存期(HR = 0.74;95% CI:0.59–0.92)及确认的客观缓解率(44.3% vs. 29.1%)方面也显示出统计学显著差异。任意级别的药物相关不良事件发生率分别为93.0%(曲妥珠单抗德鲁司他肯组)和91.4%(雷莫芦单抗联合紫杉醇组);3级或以上不良事件的发生率分别为50.0%和54.1%。由独立评估委员会认定的药物相关间质性肺病或肺炎在曲妥珠单抗德鲁司他肯组为13.9%(其中33例为1或2级,1例为3级),在对照组为1.3%(2例为3级,1例为5级)。
结论:在HER2阳性转移性胃癌或胃食管结合部腺癌患者中,曲妥珠单抗德鲁司他肯较雷莫芦单抗联合紫杉醇可显著延长总生存期。两组中不良事件均较常见。曲妥珠单抗德鲁司他肯相关的间质性肺病或肺炎虽为既知风险,且发生率较高,但多为低等级事件。(本研究由第一三共公司和阿斯利康资助;DESTINY-Gastric04试验注册号:NCT04704934)
Tarlatamab in Small-Cell Lung Cancer after Platinum-Based Chemotherapy
Tarlatamab用于铂类化疗后小细胞肺癌患者的治疗研究
美国纪念斯隆凯特琳癌症中心等机构联合发表
Abstract
Background: Tarlatamab, a bispecific delta-like ligand 3-directed T-cell engager immunotherapy, received accelerated approval for the treatment of patients with previously treated small-cell lung cancer. Whether tarlatamab is more effective than chemotherapy in the treatment of patients whose small-cell lung cancer has progressed during or after initial platinum-based chemotherapy is not known.
Methods: We conducted a multinational, phase 3, open-label trial to compare tarlatamab with chemotherapy as second-line treatment in patients with small-cell lung cancer whose disease had progressed during or after platinum-based chemotherapy. Patients were randomly assigned to receive tarlatamab or chemotherapy (topotecan, lurbinectedin, or amrubicin). The primary end point was overall survival. Key secondary end points were investigator-assessed progression-free survival and patient-reported outcomes. Results of the prespecified interim analysis (data-cutoff date, January 29, 2025) are reported.
Results: A total of 509 patients were randomly assigned to receive tarlatamab (254 patients) or chemotherapy (255 patients). Treatment with tarlatamab resulted in significantly longer overall survival than chemotherapy (median, 13.6 months [95% confidence interval {CI}, 11.1 to not reached] vs. 8.3 months [95% CI, 7.0 to 10.2]; stratified hazard ratio for death, 0.60; 95% CI, 0.47 to 0.77; P<0.001). Tarlatamab treatment also had a significant benefit with respect to progression-free survival and cancer-related dyspnea and cough as compared with chemotherapy. The incidence of adverse events of grade 3 or higher was lower with tarlatamab than with chemotherapy (54% vs. 80%), as was the incidence of adverse events resulting in treatment discontinuation (5% vs. 12%).
Conclusions: Treatment with tarlatamab led to longer overall survival than chemotherapy among patients with small-cell lung cancer whose disease had progressed during or after platinum-based chemotherapy. (Funded by Amgen; DeLLphi-304 ClinicalTrials.gov number, NCT05740566.)
摘要
背景:Tarlatamab是一种双特异性、靶向Delta-like ligand 3(DLL3)的T细胞接合免疫治疗药物,已获得加速批准,用于治疗既往接受过治疗的小细胞肺癌患者。然而,Tarlatamab是否优于化疗,用于治疗在初始铂类化疗期间或之后进展的小细胞肺癌患者,尚不明确。
方法:我们开展了一项多国、多中心、开放标签的3期临床试验,比较Tarlatamab与化疗在小细胞肺癌患者中的二线治疗效果。这些患者的疾病在接受过铂类化疗期间或之后发生进展。患者被随机分配接受Tarlatamab或化疗(拓扑替康、鲁比替丁或阿曲霉素)。主要终点为总生存期。关键次要终点包括研究者评估的无进展生存期和患者报告的结局。本报告为预先设定的中期分析结果(数据截止日期:2025年1月29日)。
结果:共有509名患者被随机分为Tarlatamab组(254人)和化疗组(255人)。Tarlatamab治疗使总生存期显著延长(中位生存期为13.6个月 [95%置信区间:11.1个月至未达到],对比化疗组的8.3个月 [95%置信区间:7.0至10.2个月];死亡的分层风险比为0.60 [95%置信区间:0.47至0.77],P<0.001)。与化疗相比,Tarlatamab在无进展生存期和与癌症相关的呼吸困难及咳嗽方面也表现出显著优势。3级或更高级别不良事件的发生率在Tarlatamab组中低于化疗组(54% vs. 80%),因不良事件而停药的比例也较低(5% vs. 12%)。
结论:在小细胞肺癌患者中,对于那些在铂类化疗期间或之后病情进展者,Tarlatamab治疗相比化疗能显著延长总生存期。(本研究由安进公司资助;DeLLphi-304临床试验注册号:NCT05740566。)
Ivermectin to Control Malaria - A Cluster-Randomized Trial
伊维菌素控制疟疾:一项集群随机对照试验
西班牙全球健康研究所等机构联合发表
Abstract
Background: Malaria control and elimination is threatened by the spread of insecticide resistance and behavioral adaptation of vectors. Whether mass administration of ivermectin, a broad-spectrum antiparasitic drug that also kills mosquitoes feeding on treated persons, can reduce malaria transmission is unclear.
Methods: We conducted a cluster-randomized trial in Kwale, a county in coastal Kenya in which malaria is highly endemic and coverage and use of insecticide-treated nets are high. Clusters of household areas were randomly assigned in a 1:1 ratio to receive mass administration of ivermectin (400 [mu]g per kilogram of body weight) or albendazole (400 mg, active control) once a month for 3 consecutive months at the beginning of the "short rains" season. Children 5 to 15 years of age were tested for malaria infection monthly for 6 months after the first round of treatment. The two primary outcomes were the cumulative incidence of malaria infection (assessed among children 5 to 15 years of age) and of adverse events (assessed among all eligible participants). Analyses were performed with generalized estimating equations in accordance with the intention-to-treat principle.
Results: A total of 84 clusters comprising 28,932 eligible participants underwent randomization. The baseline characteristics of the participants were similar in the trial groups. Six months after the first round of treatment, the incidence of malaria infection was 2.20 per child-year at risk in the ivermectin group and 2.66 per child-year at risk in the albendazole group; the adjusted incidence rate ratio (ivermectin vs. albendazole) was 0.74 (95% confidence interval [CI], 0.58 to 0.95, P=0.02). The incidence of serious adverse events per 100 treatments did not differ significantly between the trial groups (incidence rate ratio, 0.63; 95% CI, 0.21 to 1.91).
Conclusions: Among children 5 to 15 years of age who were living in an area with high coverage and use of bed nets, ivermectin, administered once a month for 3 consecutive months, resulted in a 26% lower incidence of malaria infection than albendazole. No safety concerns were identified. (Funded by Unitaid; BOHEMIA ClinicalTrials.gov number, NCT04966702; Pan African Clinical Trial Registry number, PACTR202106695877303.)
摘要
背景:杀虫剂抗性的发展和蚊媒行为的适应性改变威胁着疟疾的控制和消除。伊维菌素是一种广谱抗寄生虫药物,能够杀死叮咬受治疗者的蚊子,但其大规模使用是否可以降低疟疾传播尚不明确。
方法:我们在肯尼亚沿海地区Kwale县开展了一项集群随机对照试验,该地区疟疾高度流行,且杀虫剂处理蚊帐的覆盖率和使用率较高。研究将家庭区域按1:1比例随机分配至接受伊维菌素(每公斤体重400微克)或阿苯达唑(400毫克,作为活性对照)的大规模用药,每月一次,连续三个月,起始时间为“短雨季”的开始。5至15岁儿童在首次用药后连续6个月每月检测一次疟疾感染。主要研究终点为:该年龄段儿童疟疾感染的累积发生率,以及所有合格参与者的不良事件发生率。分析采用广义估计方程,按意向性治疗原则进行。
结果:共有84个集群、28,932名符合条件的参与者完成随机分组。两组受试者的基线特征相似。首次用药6个月后,伊维菌素组的疟疾感染发生率为每儿童每年2.20例,阿苯达唑组为2.66例;调整后的发病率比为0.74(95%置信区间[CI]:0.58至0.95,P=0.02)。每100次治疗所产生的严重不良事件发生率在两组之间无显著差异(发病率比为0.63;95% CI:0.21至1.91)。
结论:在高蚊帐覆盖率和使用率地区的5至15岁儿童中,伊维菌素每月一次、连续三个月的用药方案可使疟疾感染发生率降低26%,且未发现安全性问题。(本研究由Unitaid资助;BOHEMIA研究注册号:NCT04966702,Pan African注册号:PACTR202106695877303。)
第393卷第5期一共发表34篇内容,其中原始研究3篇。
Measurable Residual Disease-Guided Therapy in Newly Diagnosed Myeloma
可测量残留病指导的新诊断多发性骨髓瘤治疗策略
法国骨髓瘤研究组等机构联合发表
Abstract
Background: Measurable residual disease (MRD) is a major prognostic factor in newly diagnosed multiple myeloma. An assessment of an MRD-guided consolidation strategy in patients who are eligible for autologous stem-cell transplantation (ASCT) may be useful.
Methods: In this phase 3 trial, we randomly assigned transplantation-eligible patients with newly diagnosed myeloma who had completed induction therapy with isatuximab, carfilzomib, lenalidomide, and dexamethasone (Isa-KRd) to receive consolidation therapy according to their MRD status. Patients who were MRD-negative at 10-5 sensitivity (i.e.,<1 cancer cell per 100,000 normal cells, as assessed by next-generation sequencing) were assigned to undergo ASCT and receive Isa-KRd for two cycles (ASCT group) or to receive Isa-KRd for six cycles (Isa-KRd group). Patients who were MRD-positive at 10-5 sensitivity were assigned to undergo tandem ASCT (two ASCTs within a short period; tandem ASCT group) or to undergo ASCT and receive Isa-KRd for two cycles (single ASCT group). The primary end point was an MRD-negative status at 10-6 sensitivity before maintenance therapy.
Results: Among 485 patients who were MRD-negative at 10-5 sensitivity after induction, a premaintenance MRD-negative status at 10-6 sensitivity occurred in 86% in the ASCT group and in 84% in the Isa-KRd group (adjusted relative risk, 1.02; 95% confidence interval [CI], 0.95 to 1.10; P=0.64). Among 233 patients who were MRD-positive at 10-5 sensitivity after induction, a premaintenance MRD-negative status at 10-6 sensitivity occurred in 32% in the tandem ASCT group and in 40% in the single ASCT group (adjusted relative risk, 0.82; 95% CI, 0.58 to 1.15; P=0.31); 15% of the patients in the tandem ASCT group did not undergo a second ASCT. During consolidation, disease progression occurred in 5 patients and death unrelated to disease progression occurred in 2 patients - all were in the Isa-KRd or tandem ASCT groups. No new safety signals were observed. The median follow-up was 16.8 months in the ASCT and Isa-KRd groups and 16.3 months in the tandem ASCT and single ASCT groups.
Conclusions: Among patients who were MRD-negative at 10-5 sensitivity after induction, the percentage with a premaintenance MRD-negative status at 10-6 sensitivity was not significantly higher with ASCT than with Isa-KRd. Among patients who were MRD-positive status at 10-5 sensitivity after induction, the percentage with a premaintenance MRD-negative status at 10-6 sensitivity was not significantly higher with tandem ASCT than with single ASCT. (Funded by Intergroupe Francophone du Myelome and others; MIDAS ClinicalTrials.gov number, NCT04934475.)
摘要
背景:可测量残留疾病(MRD)是新诊断的多发性骨髓瘤的一个重要预后因素。对符合自体干细胞移植(ASCT)条件的患者进行以 MRD 指导的巩固治疗策略的评估可能很有意义。
方法:在这项III期临床试验中,作者将完成Isatuximab、Carfilzomib、Lenalidomide和Dexamethasone(Isa-KRd)诱导治疗且适合移植的新诊断多发性骨髓瘤患者随机分配到根据其MRD状态接受巩固治疗的组别。在诱导治疗后MRD阴性(10⁻⁵敏感度,即每10万正常细胞中少于1个癌细胞,通过下一代测序评估)的患者被分配到接受ASCT并进行两周期Isa-KRd治疗(ASCT组)或仅接受六周期Isa-KRd治疗(Isa-KRd组)。在诱导治疗后MRD阳性(10⁻⁵敏感度,即每10万正常细胞中≥1个癌细胞)的患者被分配到接受串联ASCT(短时间内两次ASCT;串联ASCT组)或接受ASCT并进行两周期Isa-KRd治疗(单次ASCT组)。主要终点是在维持治疗前达到10⁻⁶敏感度(每百万正常细胞中少于1个癌细胞)的MRD阴性状态。
结果:在485名诱导治疗后MRD阴性(10⁻⁵敏感度)的患者中,ASCT组中有86%的患者在维持治疗前达到10⁻⁶敏感度的MRD阴性状态,Isa-KRd组为84%(调整后相对风险1.02;95%置信区间[CI],0.95至1.10;P=0.64)。在233名诱导治疗后MRD阳性(10⁻⁵敏感度)的患者中,串联ASCT组有32%的患者在维持治疗前达到10⁻⁶敏感度的MRD阴性状态,单次ASCT组为40%(调整后相对风险0.82;95% CI,0.58至1.15;P=0.31);串联ASCT组中有15%的患者未进行第二次ASCT。在巩固治疗期间,5名患者出现疾病进展,2名患者出现与疾病进展无关的死亡——这些患者均来自Isa-KRd组或串联ASCT组。未观察到新的安全信号。ASCT和Isa-KRd组的中位随访时间为16.8个月,串联ASCT和单次ASCT组为16.3个月。
结论:在诱导治疗后MRD阴性(10⁻⁵敏感度)的患者中,ASCT组与Isa-KRd组相比,在维持治疗前达到10⁻⁶敏感度的MRD阴性状态的比例并无显著差异。在诱导治疗后MRD阳性(10⁻⁵敏感度)的患者中,串联ASCT组与单次ASCT组相比,在维持治疗前达到10⁻⁶敏感度的MRD阴性状态的比例也无显著差异。(由法国骨髓瘤研究组和其他机构资助;MIDAS临床试验注册号为NCT04934475。)
Mitochondrial Donation and Preimplantation Genetic Testing for mtDNA Disease
线粒体捐赠和植入前遗传检测在线粒体DNA疾病中的应用
英国线粒体研究联盟等机构联合发表
Abstract
Background: Children born to women who carry pathogenic variants in mitochondrial DNA (mtDNA) are at risk for a range of clinical syndromes collectively known as mtDNA disease. Mitochondrial donation by pronuclear transfer involves transplantation of nuclear genome from a fertilized egg from the affected woman to an enucleated fertilized egg donated by an unaffected woman. Thus, pronuclear transfer offers affected women the potential to have a genetically related child with a reduced risk of mtDNA disease.
Methods: We offered mitochondrial donation (by pronuclear transfer) or preimplantation genetic testing (PGT) to a series of women with pathogenic mtDNA variants who sought to reduce the transmission of these variants to their children. Patients with heteroplasmy (variants present in a proportion of copies of mtDNA) were offered PGT, and patients with homoplasmy (variants present in all copies of mtDNA) or elevated heteroplasmy were offered pronuclear transfer.
Results: Clinical pregnancies were confirmed in 8 of 22 patients (36%) and 16 of 39 patients (41%) who underwent an intracytoplasmic sperm injection procedure for pronuclear transfer or for PGT, respectively. Pronuclear transfer resulted in 8 live births and 1 ongoing pregnancy. PGT resulted in 18 live births. Heteroplasmy levels in the blood of the 8 infants whose mothers underwent pronuclear transfer ranged from undetectable to 16%. Levels of the maternal pathogenic mtDNA variant were 95 to 100% lower in 6 newborns and 77 to 88% lower in 2 newborns than in the corresponding enucleated zygotes. Heteroplasmy levels were known for 10 of the 18 infants whose mothers underwent PGT and ranged from undetectable to 7%.
Conclusions: We found that mitochondrial donation through pronuclear transfer was compatible with human embryo viability. An integrated program involving pronuclear transfer and PGT was effective in reducing the transmission of homoplasmic and heteroplasmic pathogenic mtDNA variants. (Funded by NHS England and others.)
摘要
背景:携带线粒体DNA(mtDNA)致病变异的女性所生的子女有患一系列临床综合征的风险,这些综合征统称为mtDNA疾病。原核移植的线粒体捐赠涉及将受影响女性的受精卵的核基因组移植到未受影响女性捐赠的去核受精卵中。因此,原核移植为受影响的女性提供了一个生育遗传相关但患病风险降低的子女的潜在机会。
方法:作者为一系列携带致病变异的女性提供了线粒体捐赠(通过原核移植)或植入前遗传检测(PGT),这些女性希望减少将这些变异传给子女的风险。患有异质性(mtDNA部分拷贝中存在变异)的患者被提供PGT,而患有同质性(所有mtDNA拷贝中均存在变异)或高异质性的患者被提供原核移植。
结果:在接受原核移植或PGT的患者中,22名患者中有8名(36%)和39名患者中有16名(41%)通过单精子注射程序确认了临床妊娠。原核移植导致8例活产和1例正在进行的妊娠。PGT导致18例活产。接受原核移植的8名婴儿的血液中异质性水平从无法检测到16%不等。与对应的去核合子相比,6名新生儿的母系致病变异mtDNA水平降低了95%至100%,2名新生儿降低了77%至88%。接受PGT的18名婴儿中,已知10名婴儿的异质性水平,范围从无法检测到7%。
结论:作者发现通过原核移植的线粒体捐赠与人类胚胎的存活性相容。一个涉及原核移植和PGT的综合项目在减少同质性和异质性致病变异的传播方面是有效的。(由NHS英格兰和其他机构资助)
A Randomized Trial of Acute Normovolemic Hemodilution in Cardiac Surgery
心脏手术中急性等容性血液稀释的随机对照试验
意大利米兰IRCCS San Raffaele科学研究所等机构联合发表
Abstract
Background: Patients undergoing cardiac surgery often receive red-cell transfusions, along with the associated risks and costs. Early intraoperative normovolemic hemodilution (i.e., acute normovolemic hemodilution [ANH]) is a blood-conservation technique that entails autologous blood collection before initiation of cardiopulmonary bypass and reinfusion of the collected blood after bypass weaning. More data are needed on whether ANH reduces the number of patients receiving allogeneic red-cell transfusion.
Methods: In a multinational, single-blind trial, we randomly assigned adults from 32 centers and 11 countries who were undergoing cardiac surgery with cardiopulmonary bypass to receive ANH (withdrawal of >=650 ml of whole blood with crystalloids replacement if needed) or usual care. The primary outcome was the transfusion of at least one unit of allogeneic red cells during the hospital stay. Secondary outcomes were death from any cause within 30 days after surgery or during the hospitalization for surgery, bleeding complications, ischemic complications, and acute kidney injury.
Results: A total of 2010 patients underwent randomization; 1010 were assigned to ANH and 1000 to usual care. Among patients with available data, 274 of 1005 (27.3%) in the ANH group and 291 of 997 (29.2%) in the usual-care group received at least one allogeneic red-cell transfusion (relative risk, 0.93; 95% confidence interval, 0.81 to 1.07; P=0.34). Surgery for postoperative bleeding was performed in 38 of 1004 patients (3.8%) in the ANH group and 26 of 995 patients (2.6%) in the usual-care group. Death within 30 days or during hospitalization occurred in 14 of 1008 patients (1.4%) in the ANH group and 16 of 997 patients (1.6%) in the usual-care group. Safety outcomes were similar in the two groups.
Conclusions: Among adults undergoing cardiac surgery, ANH did not reduce the number of patients receiving allogeneic red-cell transfusion. (Funded by the Italian Ministry of Health; ANH ClinicalTrials.gov number, NCT03913481.)
摘要
背景:接受心脏手术的患者常需要接受红细胞输血,这带来了相关的风险和成本。早期术中等容性血液稀释(即急性等容性血液稀释 [ANH])是一种血液保护技术,涉及在体外循环开始前采集患者自身的血液,并在体外循环结束后重新输回患者体内。目前尚缺乏关于ANH是否能减少接受异体红细胞输血患者数量的更多数据。
方法:在一项多国、单盲试验中,我们将来自11个国家32个中心的成年患者随机分配到接受ANH(抽取≥650ml全血,并根据需要使用晶体液替代)或常规治疗组。这些患者均需接受体外循环下的心脏手术。主要结果是住院期间接受至少一单位异体红细胞输血。次要结果包括术后30天内或住院期间的任何原因死亡、出血并发症、缺血并发症和急性肾损伤。
结果:共有2010名患者接受了随机分配,其中1010名分配到ANH组,1000名分配到常规治疗组。在有可用数据的患者中,ANH组1005名患者中有274名(27.3%)和常规治疗组997名患者中有291名(29.2%)接受了至少一单位的异体红细胞输血(相对风险0.93,95%置信区间0.81至1.07,P=0.34)。ANH组1004名患者中有38名(3.8%)和常规治疗组995名患者中有26名(2.6%)因术后出血接受了手术。ANH组1008名患者中有14名(1.4%)和常规治疗组997名患者中有16名(1.6%)在术后30天内或住院期间死亡。两组的安全性结果相似。
结论:在接受心脏手术的成年人中,ANH并未减少接受异体红细胞输血的患者数量。(由意大利卫生部资助;ANH ClinicalTrials.gov编号,NCT03913481。)

汇报人:徐凡淇
审核:张子妍、任建君
