



精读分享│:EZH2通过激活整合素β1-FAK通路参与TGF β信号传导从而促进乳腺癌骨转移
英文题目:EZH2 engages TGFβ signaling to promote breast cancer bone metastasis via integrin β1-FAK activation
中文题目:EZH2通过激活整合素β1-FAK通路参与TGF β信号传导从而促进乳腺癌骨转移
期刊:Nature communications(IF=17.69)

Abstract
Bone metastases occur in 50–70% of patients with late-stage breast cancers and effective therapies are needed. The expression of enhancer of zeste homolog 2 (EZH2) is correlated with breast cancer metastasis, but its function in bone metastasis hasn’t been well-explored. Here we report that EZH2 promotes osteolytic metastasis of breast cancer through regulating transforming growth factor beta (TGFβ) signaling. EZH2 induces cancer cell proliferation and osteoclast maturation, whereas EZH2 knockdown decreases bone metastasis incidence and outgrowth in vivo. Mechanistically, EZH2 transcriptionally increases ITGB1, which encodes for integrin β1. Integrin β1 activates focal adhesion kinase (FAK), which phosphorylates TGFβ receptor type I (TGFβRI) at tyrosine 182 to enhance its binding to TGFβ receptor type II (TGFβRII), thereby activating TGFβ signaling. Clinically applicable FAK inhibitors but not EZH2 methyltransferase inhibitors effectively inhibit breast cancer bone metastasis in vivo. Overall, we find that the EZH2-integrin β1-FAK axis cooperates with the TGFβ signaling pathway to promote bone metastasis of breast cancer.
摘要
骨转移发生在 50-70%的晚期乳腺癌患者中,需要有效的治疗。zeste 同源物增强子 2( EZH 2)的表达与乳腺癌的转移有关,但其在骨转移中的作用尚不清楚。在这里,我们报告 EZH 2 通过调节转化生长因子β(TGFβ)信号促进乳腺癌的溶骨性转移。EZH 2 诱导癌细胞增殖和破骨细胞成熟,而 EZH 2 敲低降低体内骨转移发生率和进展。从机制上讲,EZH 2 转录性地上调 ITGB 1,ITGB 1 编码整合素β1。整合素β1 激活粘着斑激酶(FAK),FAK 使 I 型 TGFβ受体(TGFβRI)在酪氨酸 182 处磷酸化,以增强其与 II 型 TGFβ受体(TGFβRII)的结合,从而激活 TGFβ信号传导。 临床适用的 FAK 抑制剂而非 EZH 2 甲基转移酶抑制剂在体内有效抑制乳腺癌骨转移。总之,研究发现 EZH 2-integrin β1-FAK 轴协同 TGFβ信号通路促进乳腺癌骨转移。
研究背景:
乳腺癌是全球女性个体中最常见的诊断癌症。约50-70%的晚期乳腺癌患者发生骨转移,导致骨相关事件,包括疼痛、病理性骨折、脊髓压迫、高钙血症和其他并发症。骨转移的治疗是有限的,仅仅为姑息治疗;标准的抗骨吸收药物,化疗和放疗可以延迟或减轻骨相关事件,但不能治愈骨转移。全面探讨骨转移的分子机制可能为骨转移患者提供新的治疗策略。乳腺癌骨转移常引起溶骨性病变,导致大量骨吸收和骨折。溶骨性骨吸收引起几种生长因子的分泌,包括转化生长因子β(TGFβ)。骨转移依赖于所谓的 “恶性循环”,这是指在促进不受控制的肿瘤生长和破骨细胞活性的癌细胞,成骨细胞和破骨细胞之间的前馈循环煽动。
TGFβ在癌症的发生和发展中起双重作用:它在癌前细胞中作为肿瘤抑制因子发挥作用,但通过增强上皮-间质转化、血管生成和免疫抑制诱导乳腺癌转移。研究已经充分证实,TGFβ是驱动前馈恶性循环以促进骨中转移性癌细胞生长的关键细胞因子。 在经典的 TGFβ信号传导中,活性 TGFβ与其受体 TGFβ受体 II 型(TGFβRII)结合,后者结合并激活细胞膜上的 TGFβ受体 I 型(TGFβRI)。TGFβRI 磷酸化下游信号分子 Smad 2/3,与 Smad 4 形成复合物;然后将 Smad 2/3/4 复合物移位至细胞核。核 Smad 2/3/4 复合物作为转录因子工作,以启动靶基因的转录。 非经典 TGFβ信号传导作为 Smad 非依赖性途径通过激活 p38 丝裂原活化蛋白激酶(MAPK)、细胞外信号调节激酶(ERK)、c-Jun N-末端激酶(JNK)或磷酸肌醇 3-激酶(PI 3 K)/AKT 信号传导发挥作用。
研究方法和思路:
研究结果:
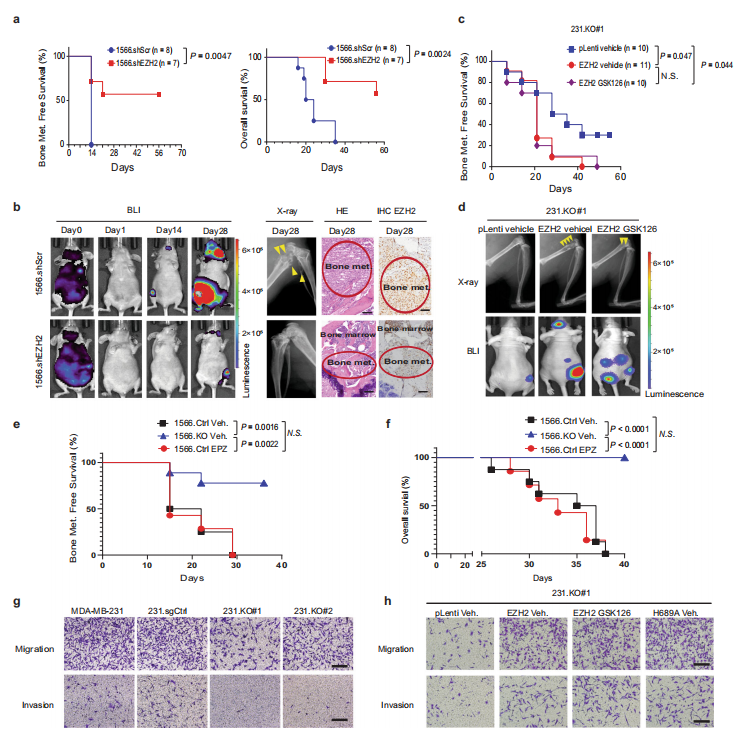
EZH2促进乳腺癌骨转移,EZH2甲基转移酶抑制剂不能阻止这一过程
作者用shRNA转染细胞,构建了EZH2敲除细胞系及其对照细胞系,再将细胞注射到裸鼠左心室,从图a可以看出,EZH2敲除的小鼠无骨转移生存期和总生存期都更长。图b用生物发光成像、X射线成像和HE染色表示小鼠体内骨转移情况,敲除EZH2的小鼠发生骨转移比对照组少。图c将EZH2敲除小鼠用载体处理或使其重新表达野生型EZH2,再用载体或GSK126处理有EZH2表达的细胞,GSK126是一种小分子EZH2甲基转移酶抑制剂,结果发现重新表达EZH2的小鼠生存期下降,GSK126处理的EZH2敲除小鼠与对照组相比生存期变化不明显。在图d的生物发光成像和X射线成像结果中,EZH2重新表达增加了骨转移,同样,GSK126并不能抑制这种作用。从图e和f的结果来看,敲除EZH2的细胞生存期延长,而另一种甲基转移酶抑制剂EPZ6438与GSK126一样,无法抑制EZH2诱导的小鼠生存期下降。以上结果都说明,EZH2诱导小鼠生存期下降和促进骨转移的作用不依赖于甲基转移酶。接着,作者比较了EZH2对肿瘤细胞迁移和侵袭的影响。从图g来看,敲除EZH2的肿瘤细胞发生迁移和侵袭的能力与对照组相比会更弱。图h表示的是,EZH2和甲基转移酶死亡突变体H689A处理会增加细胞的迁移和侵袭;与此同时,GSK126并不会抑制受EZH2诱导增加的细胞的迁移和侵袭能力。因此,EZH2会促进癌细胞迁移和侵袭,但这一功能可能并不依赖于EZH2的甲基转移酶。
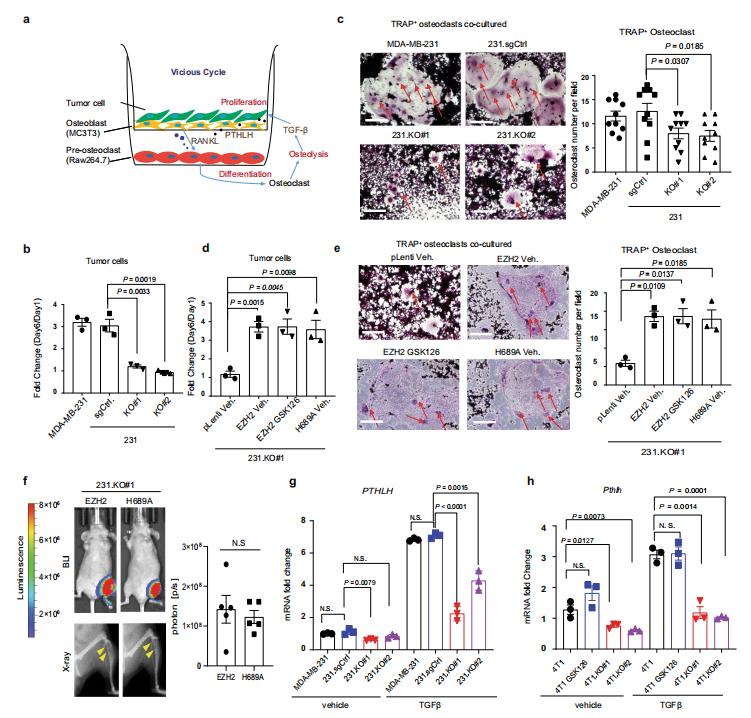
EZH2调节乳腺癌骨转移的恶性循环
图a展示了在TGFβ刺激下,将乳腺癌细胞与破骨前细胞和成骨细胞三重共培养,模拟乳腺癌骨转移微环境的恶性循环。从图b显示,敲除了EZH2的肿瘤细胞生长被抑制。同时,图c的结果揭示了,敲除EZH2会使得成熟破骨细胞变少。为了检测EZH2甲基转移酶在乳腺癌骨转移恶性循环中的作用,图d中敲除EZH2的肿瘤细胞再用H689A(EZH2甲基转移酶死亡突变体)处理之后,与野生型EZH2处理类似,细胞生长增殖和破骨细胞的成熟增加。图e中重新表达野生型EZH2或用H689A处理的细胞,细胞生长增殖更多,破骨细胞成熟也增加;再经过GSK126处理,这种效果也没有发生改变。图f将H689A细胞导入EZH2敲除的小鼠体内,通过生物发光成像和X射线成像观察小鼠体内骨转移,发现H689A与EZH2一样,都会诱导小鼠发生骨转移。总之,EZH2会促进乳腺癌骨转移,EZH2甲基转移酶抑制剂和H689A甲基转移酶死亡突变都不能阻断这一恶性循环。另外,PTHLH是乳腺癌骨转移的重要介质,从图g和h可以看出,敲除EZH2可以降低PTHLH mRNA表达,减少PTHLH分泌到骨微环境中去激活骨溶解。
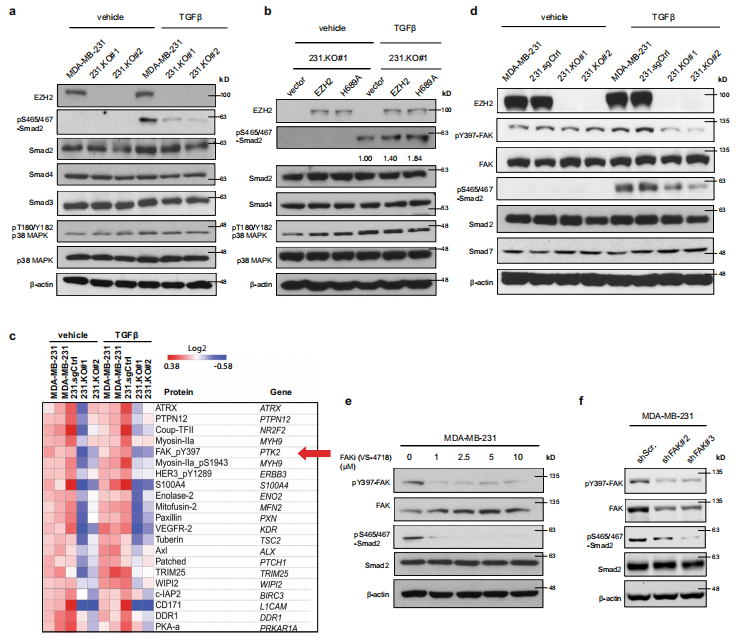
EZH2在TGF β刺激下增加pS465/467-Smad2和pY397-FAK水平
PTHLH是TGFβ下游基因,受磷酸化Smad2转录因子调控,从图a可以看出,在TGFβ刺激下,敲除EZH2会抑制磷酸化Smad2水平,继而减少PTHLH分泌。图b在TGFβ的刺激下,敲除EZH2后重新表达野生型EZH2或H689A突变体,磷酸化Smad2的水平升高了。这两个实验说明EZH2甲基转移酶确实参与调控Smad2磷酸化。接着,作者用反相蛋白阵列RPPA比较正常肿瘤细胞和EZH2敲除的肿瘤细胞在有或没有TGF β刺激时的蛋白变化情况。图c热图结果表明敲除EZH2使FAK上397位点酪氨酸磷酸化明显减少了。图d的结果表明,敲除EZH2的细胞在TGF β刺激下,FAK和Smad2磷酸化都减少。总之,EZH2在TGF β刺激下激活FAK和Smad2信号通路。在图e中,作者将细胞用不同浓度的FAK抑制剂处理,再经TGF β刺激,发现当FAKi浓度越来越大,Smad2磷酸化越来越少。图f用shRNA沉默FAK时,得到了与图e相同的结论,Smad2磷酸化受到抑制。这些数据表明,EZH2先激活FAK,继而增加乳腺癌细胞中Smad2磷酸化,并激活了TGF β/ Smad2/PTHLH通路。
pY397-FAK诱导TGF βRI酪氨酸磷酸化,增强其与TGF βRII的结合
图a显示,作者通过FAK的免疫沉淀检测出FAK能与TGFβ RI结合,而不是跟TGFβ RII结合。TGF β暴露后,FAK与TGFβ I结合减少。图b结果表明FAK抑制剂使FAK与TGFβ RI的结合降低。图c展示了在MDA-MB-231细胞中抑制FAK后,用TGFβ处理并收集细胞裂解液,接着通过免疫沉淀TGFβRI并进行Western blot分析,发现FAKi显著降低了I型TGFβ受体与II型TGFβ受体的结合。接着,为了研究FAK是否可以磷酸化I型TGFβ受体,从转染外源flag标记的野生型TGFβ RI的细胞中沉淀TGFβ RI,然后用抗磷酸化酪氨酸抗体进行WB,从图d来看,FAK抑制使得I型TGFβ受体上酪氨酸磷酸化减少。因此,FAK能促进TGFβRI磷酸化,并增强与II型TGFβ受体的结合。
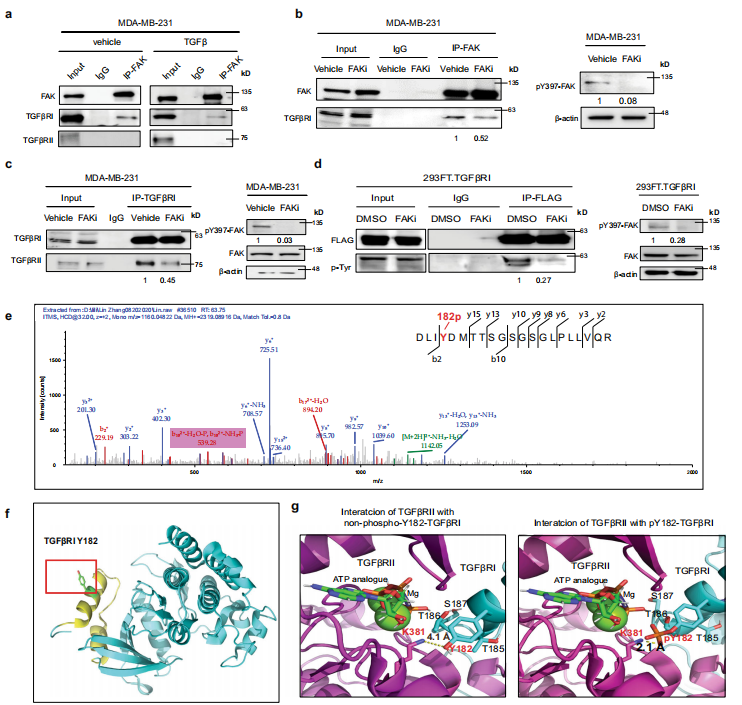
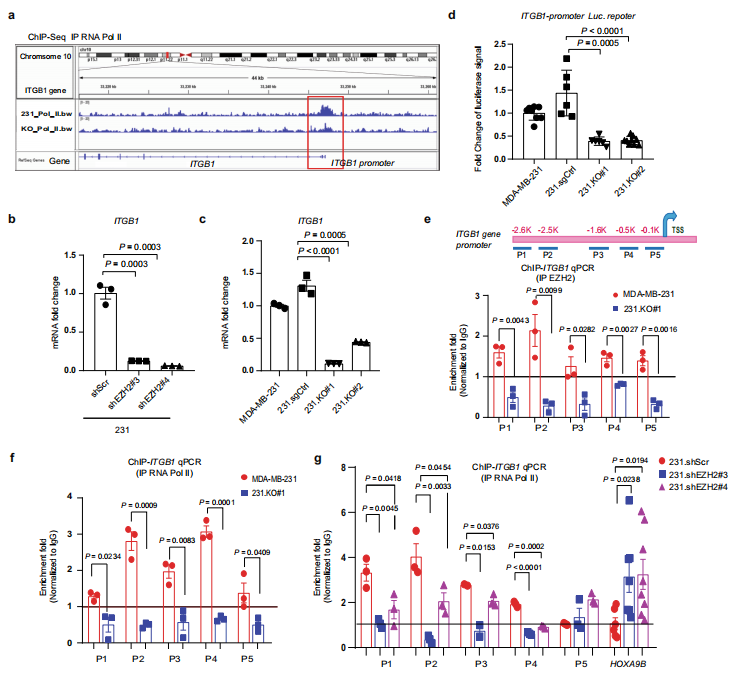
5.EZH2增加FAK上游ITGB1的表达
由于以前报道过EZH2可以作为RNA Pol II的转录辅助因子上调mRNA转录,因此作者检测了EZH2是否也可以增加编码β1的ITGB1的RNA Pol II转录。图a中的染色质免疫沉淀实验结果显示,EZH2敲除导致RNA Pol II与编码整合素的ITGB1启动子结合显著降低。图b和c的qRT-PCR结果说明沉没或敲除EZH2之后,ITGB1 mRNA表达减少。图d展示了ITGB1启动子的双荧光素酶报告基因检测结果,表明EZH2敲除细胞中ITGB1启动子活性降低。图e展示了在MDA-MB-231和231.KO#1细胞中,免疫沉淀出EZH2,并使用针对ITGB1基因启动子不同区域的PCR引物进行qPCR,检测细胞内与ITGB1结合的EZH2。作者发现,敲除EZH2之后,与ITGB1启动子的这些位点结合的EZH2确实减少了。同样的,图f在细胞中免疫沉淀出RNA Pol II之后,再用qPCR检测ITGB I与RNA Pol II的结合情况,发现EZH2敲除后,RNA Pol II与ITGB1启动子的结合也减少。图g用沉没了EZH2的细胞进行实验,与对照组相比,沉没EZH2之后,ITGB1启动子区域与RNA Pol II的结合降低了。因此,EZH2和RNA Pol II都结合在ITGB1的相同启动子区域,EZH2可能作为RNA Pol II的辅助因子,独立于其甲基转移酶功能上调ITGB1的转录。
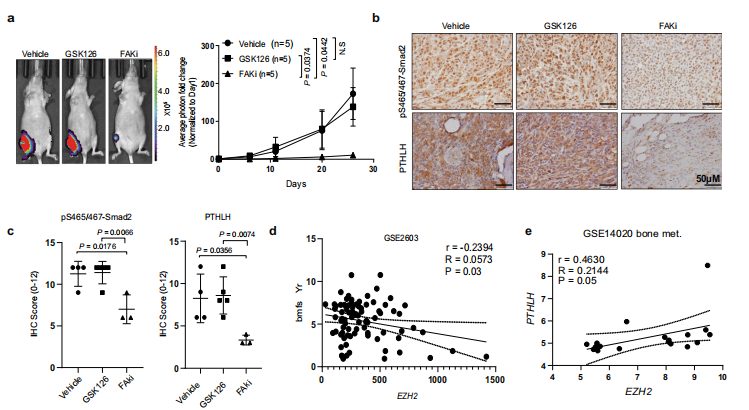
6.临床应用的FAK抑制剂可以阻断EZH2诱导的乳腺癌骨转移
图a生物发光图像的结果显示EZH2甲基转移酶抑制剂GSK126无法有效阻止骨肿瘤生长,而FAKi能够显著抑制骨肿瘤的生长。图b免疫组化以及图c的量化结果显示,FAKi能降低Smad2磷酸化和PTHLH表达,而GSK126效果不明显甚至没有效果。进一步,从图d和e可以看出,EZH2表达越高,乳腺癌患者的无骨转移生存期越短,而PTHLH水平增高,加速骨溶解。因此,FAKi能够有效阻断EZH2诱导的乳腺癌骨转移。
讨论:
本文揭示了EZH2促进乳腺癌骨转移的机制,EZH2和TGFβ信号通路通过一种不依赖于甲基转移酶的机制促进乳腺癌骨转移,其中FAK是EZH2的下游效应因子。EZH2作为RNA Pol II的转录辅助因子,增加编码整合素β1的ITGB1的转录,从而诱导FAK磷酸化。激活的FAK又磷酸化I型TGFβ受体,增加TGFβRI与TGFβRII结合,从而触发Smad2磷酸化,继而诱导PTHLH,加速骨溶解,导致更多TGFβ释放,形成恶性循环。因此,FAKi联合标准的抗骨吸收药物、化疗或放疗可能为乳腺癌骨转移患者提供额外的好处。

汇报人:王心宇
审核:张子妍、徐凡淇、任建君
