


精读分享│:从单细胞转录组解析人类肿瘤的拷贝数与克隆亚结构
英文题目:Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes
中文题目:从单细胞转录组解析人类肿瘤的拷贝数与克隆亚结构
期刊:Nature Biotechnology(IF=41.7)

Abstract
Single-cell transcriptomic analysis is widely used to study human tumors. However, it remains challenging to distinguish normal cell types in the tumor microenvironment from malignant cells and to resolve clonal substructure within the tumor. To address these challenges, we developed an integrative Bayesian segmentation approach called copy number karyotyping of aneuploid tumors (CopyKAT) to estimate genomic copy number profiles at an average genomic resolution of 5 Mb from read depth in high-throughput single-cell RNA sequencing (scRNA-seq) data. We applied CopyKAT to analyze 46,501 single cells from 21 tumors, including triple-negative breast cancer, pancreatic ductal adenocarcinoma, anaplastic thyroid cancer, invasive ductal carcinoma and glioblastoma, to accurately (98%) distinguish cancer cells from normal cell types. In three breast tumors, CopyKAT resolved clonal subpopulations that differed in the expression of cancer genes, such as KRAS, and signatures, including epithelial-to-mesenchymal transition, DNA repair, apoptosis and hypoxia. These data show that CopyKAT can aid in the analysis of scRNA-seq data in a variety of solid human tumors.
摘要
单细胞转录组分析已成为研究人类肿瘤的重要工具。然而,如何在肿瘤微环境中准确区分正常细胞与恶性细胞,以及解析肿瘤内部的克隆亚结构,仍然是重大挑战。为解决这一问题,该研究开发了一种基于整合贝叶斯分割的分析方法——非整倍体肿瘤拷贝数核型分析(CopyKAT)。该方法利用高通量单细胞RNA测序(scRNA-seq)数据的测序深度,以约5 Mb的平均分辨率推断基因组拷贝数谱。研究将CopyKAT应用于21例肿瘤(共 46,501 个单细胞)的单细胞数据分析,包括三阴性乳腺癌、胰腺导管腺癌、间变性甲状腺癌、浸润性导管癌和胶质母细胞瘤。该方法能够以高达98%的准确率区分癌细胞与正常细胞。在三例乳腺肿瘤中,CopyKAT进一步解析了具有不同癌基因(如 KRAS)表达和功能特征的克隆亚群,这些特征包括上皮-间质转化、DNA 修复、细胞凋亡和缺氧应答等。综上,CopyKAT为多种实体瘤的scRNA-seq数据解析提供了有力工具。
研究背景
单细胞RNA测序(scRNA-seq)是解析肿瘤微环境(TME)中正常细胞类型,阐明多种人类癌症中肿瘤细胞表达程序的关键技术。随着高通量测序平台的发展,研究者可以通过微液滴体系(Drop-Seq、Indrop、10X Chromium)以及纳米孔板体系(Wafergen iCELL8、SeqWell、CelSee)等技术在一次实验中并行测序数千个单细胞,每个细胞的成本低于1美元。然而,在大规模数据分析中,如何准确区分TME中的肿瘤细胞与基质细胞及免疫细胞,仍是亟待解决的难题。
基于非整倍体拷贝数谱的识别是一种有效的区分策略。非整倍体常见于大多数人类肿瘤中(约88%),而在具有二倍体基因组的基质细胞中并不存在。以往的方法(如 inferCNV 和 HoneyBadger)已证明,可以利用RNA测序技术在较大基因组区间上推断拷贝数谱。但这些方法主要针对早期 scRNA-seq 技术设计,适用于低通量且高覆盖度的数据,而不适用于新一代高通量平台(微液滴和纳米孔板),后者通过全转录组扩增,仅在mRNA的3′或5′端以极低覆盖度进行测序。此外,既有方法无法准确定位染色体断点的具体位置,也难以基于非整倍体拷贝数谱有效区分肿瘤与正常细胞。
为克服这些限制,该研究开发了CopyKAT,并应用于多种人类肿瘤的分析,成功识别非整倍体肿瘤细胞,并解析肿瘤组织内不同亚群的克隆亚结构。
研究方法与思路:
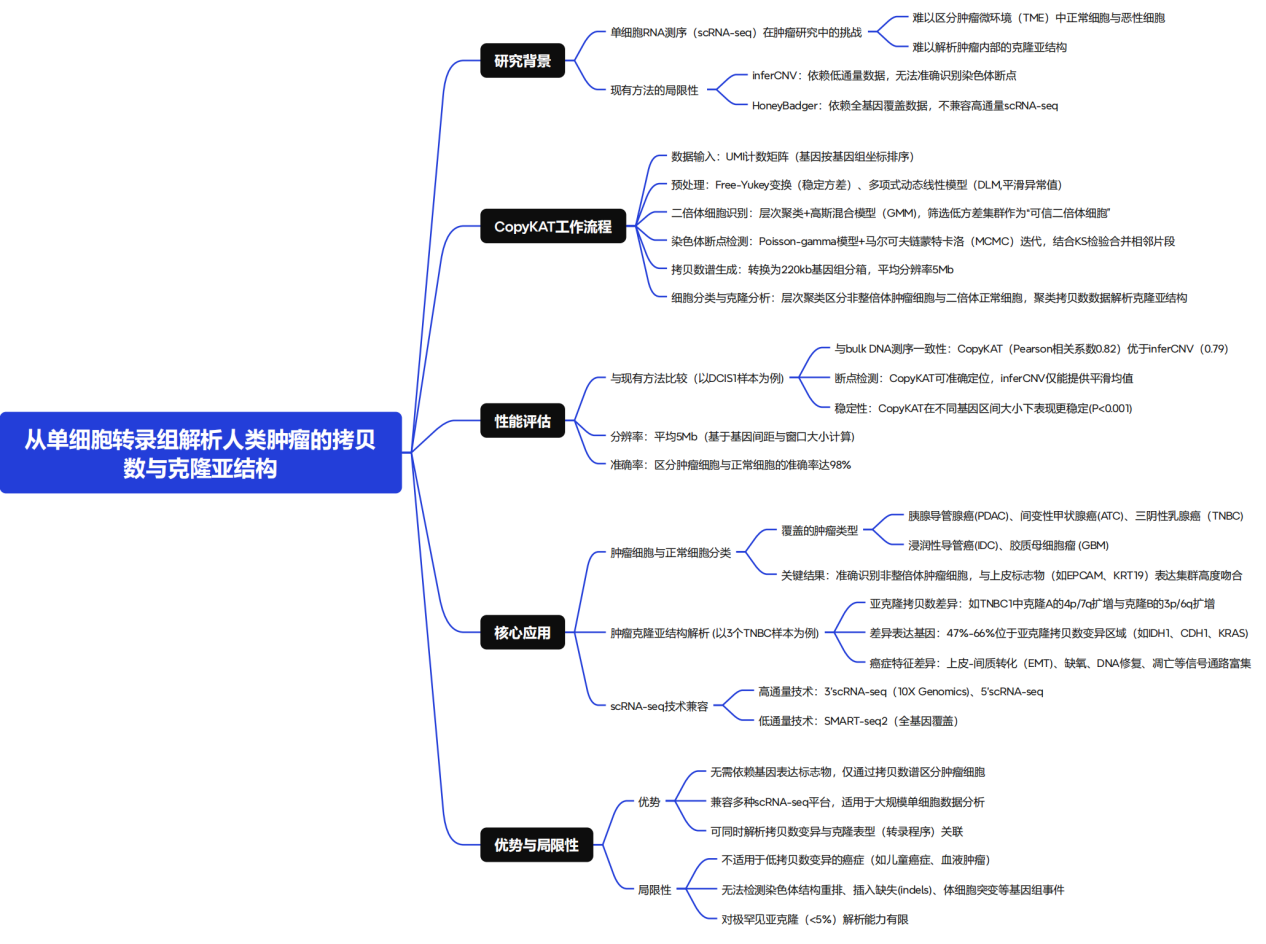
研究结果:
(1)CopyKAT 工作流程概览
CopyKAT的统计分析流程结合了贝叶斯方法与层次聚类,用于从高通量 3′ scRNA-seq 数据中计算单细胞的基因组拷贝数谱,并解析克隆亚结构(图1)。该流程以独特分子标识符(UMI)计数形成的基因表达矩阵作为输入。
分析首先对行(基因)进行基因组坐标注释和排序。随后采用Freeman–Tukey 变换来稳定方差,并通过多项式动态线性建模(DLM)平滑单细胞UMI计数中的异常值(图1a)。
接下来的步骤是检测一部分具有高度可信度的二倍体(2N)细胞,用于推断正常 2N 细胞的拷贝数基线值。为此,CopyKAT 将单细胞聚合为多个小型层次聚类,并使用高斯混合模型(GMM)估计各聚类的方差(图1b)。方差估计值最小的聚类在严格分类标准下被定义为“可信二倍体细胞”。
当样本中正常细胞数量极少,或肿瘤细胞基因组接近二倍体、仅包含有限的拷贝数异常(CNA)事件时,可能会出现误判。在这种情况下,CopyKAT 提供了一种“GMM 定义”模式,以单细胞为单位逐一识别二倍体细胞。在该模式下,假设单细胞基因表达由三个高斯分布混合而成,分别对应基因组拷贝数的增加、丢失和中性状态。当某个单细胞的中性状态基因数占所有表达基因数的 99% 以上时,该细胞即被定义为可信二倍体细胞。
为识别染色体断点,CopyKAT 首先结合 Poisson–Gamma 模型与马尔可夫链蒙特卡洛(MCMC)迭代,获得每个基因窗口的后验均值。随后采用 Kolmogorov–Smirnov(KS)检验,将相邻均值无显著差异的窗口合并(图1c)。
为提升运算效率,该研究将数千个单细胞划分为若干簇,在簇内提取共识性断点,并进一步合并,构建样本层面的统一基因组断点集合。每个细胞的最终拷贝数值则基于跨越相邻断点的所有基因的后验均值计算得到。
在此基础上,该研究将拷贝数值从基因坐标系映射至基因组位置:通过将基因重新整合为 220 kb 可变区间,生成单细胞全基因组拷贝数谱,其分辨率约为 5 Mb。该分辨率来源于基因组中位基因间距(约 20 kb)与窗口大小(25 基因)的乘积。
随后,对单细胞拷贝数谱进行层次聚类,以区分非整倍体肿瘤细胞与二倍体基质细胞;若两者基因组差异不显著,则切换至 GMM 定义模式,逐一识别肿瘤细胞(图1d)。最终,该研究对肿瘤细胞进一步聚类,识别出不同的克隆亚群,并生成其共识拷贝数谱,作为亚群基因型的代表,用于后续的表达差异分析(图1e)。
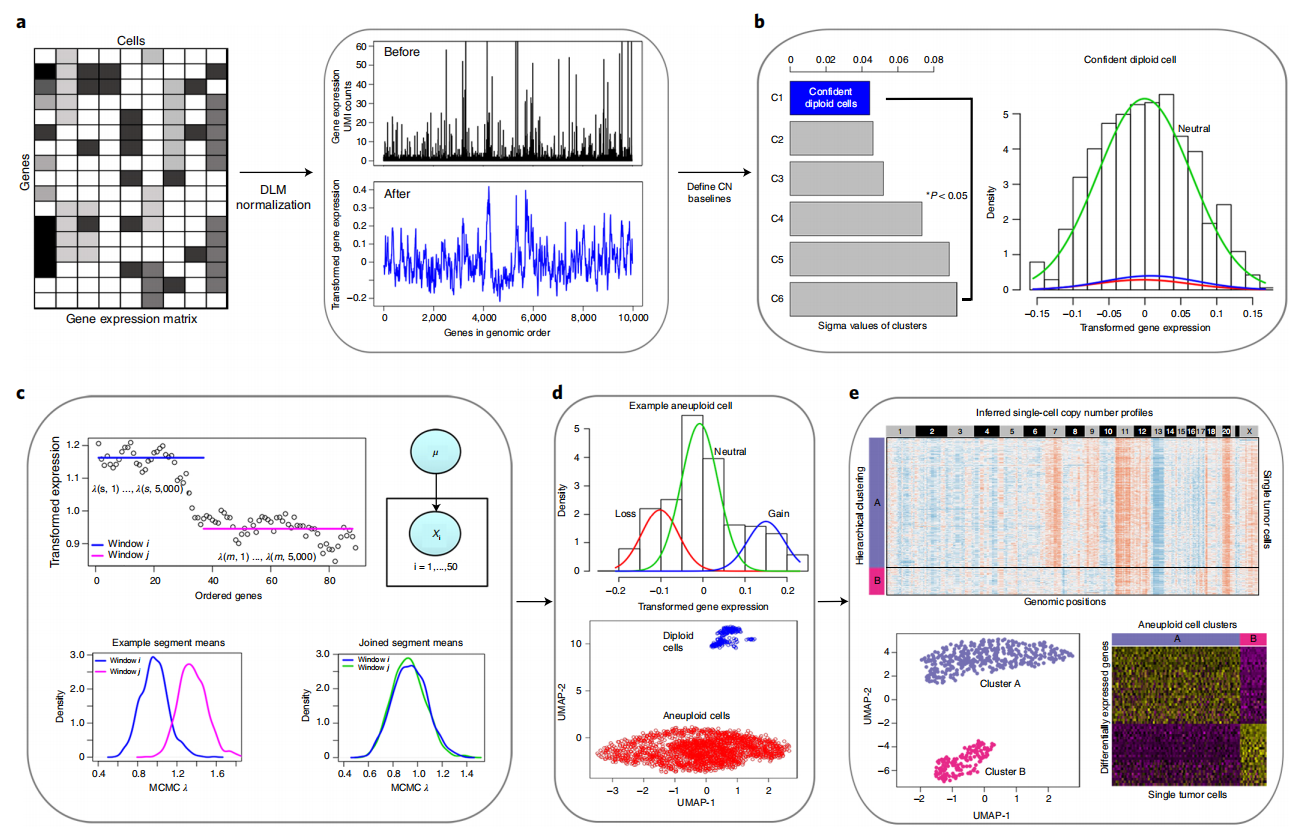
图1:CopyKAT 分析流程概览
(2)技术性能评估
为评估CopyKAT的性能,该研究对来自一例乳腺癌前期肿瘤(DCIS1)的 1,480 个单细胞肿瘤细胞进行了高通量3′scRNA 测序(10X Genomics)(图2)。该研究利用CopyKAT从scRNA-seq数据中计算全基因组拷贝数谱(图2a,b),并将结果与先前发表的方法inferCNV在相同数据上的分析结果进行比较(图2c,d)。为了生成DNA拷贝数谱的“真实参考”,该研究从DCIS1中流式分选了数百万个非整倍体肿瘤细胞,并进行了全基因组群体DNA测序(图2e,f)。结果显示,CopyKAT与群体DNA拷贝数参考谱在220-kb基因组分辨率下具有高度一致性(Pearson 相关系数=0.82)。群体DNA-seq数据中检测到的大部分主要拷贝数变异(CNA)也能在 scRNA-seq 数据中被识别,包括染色体 4、6、8、12、17、20 和 X 的扩增,以及染色体 2、3、9、10 和 15 的缺失(图2a,b)。在相同数据集上运行inferCNV时,该研究基于成纤维细胞标记基因 ACTA2 和 FN1 手动识别了基质细胞,用于提供方法所需的内部基线参考。
该研究进一步将inferCNV的结果转换为与CopyKAT相同的基因组分辨率(220-kb 可变区间),以便进行比较。虽然inferCNV的信号强度低于CopyKAT,但通过相关性分析(Pearson 相关系数 = 0.79),其结果仍与群体DNA-seq数据参考具有较高一致性(图2c,d)。然而,inferCNV的一个主要局限在于,它只能报告基因窗口的平滑平均值,无法检测染色体断点或拷贝数片段的具体坐标,而这些功能是 CopyKAT 所实现的。该研究进一步通过重复抽样不同基因数间隔的相邻局部区域,计算两种方法推断的拷贝数状态与群体 DNA-seq 拷贝数谱参考的相对距离(方法部分)。分析结果显示,CopyKAT的分割结果显著(P < 0.001,t检验)更接近参考 DNA 拷贝数状态,相比之下,inferCNV 报告的滑动窗口平均值偏离更大(图2g)。此外,CopyKAT 在不同基因窗口大小(5 至 500 个基因)下均表现出更稳定的性能(图2h)。
这些结果表明,CopyKAT 能够从高通量3′scRNA-seq数据中,以中等基因组分辨率(约5Mb)准确推断DNA拷贝数谱。
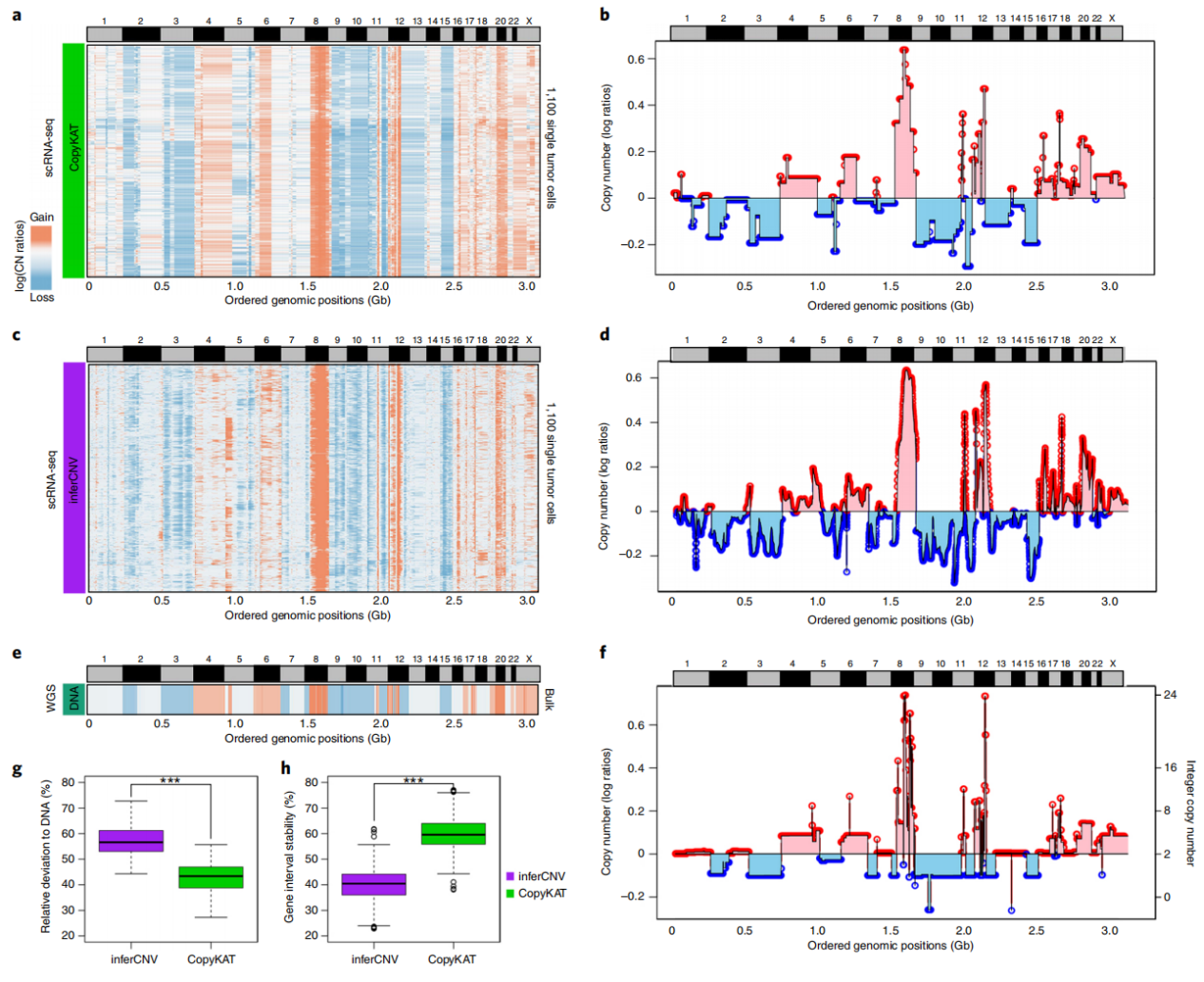
图2:群体 DNA 与单细胞 RNA 拷贝数谱比较
实体瘤中肿瘤细胞与正常细胞的分类
该研究将 CopyKAT 应用于先前发表的五例胰腺腺癌(PDAC)患者的 3′ scRNA-seq 数据,以及该研究新生成的五例三阴性乳腺癌(TNBC)和五例间变性甲状腺癌(ATC)患者的 3′scRNA-seq 数据,以基于拷贝数差异区分肿瘤细胞与正常细胞(图3)。在五例 PDAC 患者的 9,717 个单细胞转录组中,使用 CopyKAT 成功识别出每位个体中的非整倍体肿瘤细胞亚群(图3a)。预测的肿瘤细胞具有全基因组拷贝数变异(CNA),包括常见的染色体扩增 1q、3q、7p、8q、17、19 和 20,以及缺失 3p、6 和 8p,这些变化在 PDAC 中已有报道;而具有二倍体基因组的正常细胞则没有反复出现的 CNA。该研究将分类后的非整倍体肿瘤细胞投影到 UMAP 中,发现其位置与高上皮基因评分的表达簇重叠,该评分基于四个公认的肿瘤上皮标记基因(EPCAM、KRT19、KRT18 和 KRT8)计算。值得注意的是,在五例 PDAC 患者中,全基因组 CNA 仅在两类上皮簇中的一类(c1)中检测到,表明另一类簇(c2)可能为无法单凭基因表达识别的正常二倍体上皮细胞。该研究将 c1 簇指定为肿瘤细胞,因为其对应非整倍体拷贝数谱,并且 KRT19 表达水平更高,该基因是 PDAC 肿瘤中广泛使用的癌细胞标记。通过计算肿瘤细胞与 c1 表达簇的共定位情况,该研究估计 CopyKAT 在肿瘤细胞识别中具有高准确性(98.5%)。基于这些数据,该研究估计肿瘤纯度范围为 6%–18%(图3d),与以往组织病理学数据显示的结果一致,即 PDAC 患者通常因高比例基质细胞而肿瘤纯度较低。
该研究还对来自五例间变性甲状腺癌(ATC)肿瘤的 19,568 个单细胞进行了 3′高通量 scRNA-seq 测序(10X Genomics 平台)。使用 CopyKAT 分析 scRNA-seq 数据后,成功在所有五例 ATC 肿瘤中识别出非整倍体肿瘤细胞(图3b)。在推断的拷贝数数据中,检测到的常见 CNA 包括在 ATC 肿瘤中频繁报道的染色体扩增 1p、2p、5p、7、8q、11p、12、18p 和 20,以及缺失 1q、6p、13、17 和 22。在UMAP表达投影中,预测的非整倍体细胞在所有个体中对应1–2个表达簇,这些簇具有较高的上皮基因评分,其中KRT8曾被用于ATC的组织病理学识别。基于预测的非整倍体肿瘤细胞与 c1(ATC1 中的 c1 和 c2)的共定位情况,该研究估计识别肿瘤细胞的平均准确率为 97%。观察到 ATC 肿瘤的肿瘤纯度范围较广(2%–80%)(图3e),其中两例肿瘤(ATC2、ATC3)纯度较低(2%、12%),三例肿瘤(ATC1、ATC4、ATC5)纯度较高(42%–80%),与 ATC 肿瘤病理学数据显示的范围一致。
该研究进一步对五例未经治疗的三阴性乳腺癌(TNBC)患者的 8,944 个单细胞进行了 3′scRNA-seq 测序。在所有五例 TNBC 样本中,CopyKAT 成功识别出具有非整倍体和二倍体拷贝数谱的不同单细胞群体。通过聚类热图分析估计的单细胞拷贝数谱,发现了 TNBC 中已报道的常见 CNA,包括染色体扩增 1q、6p、8、10p、18p 和 Xq,以及缺失 1p、5q、17p 和 Xp。降维分析显示,预测的非整倍体肿瘤细胞对应于上皮基因评分阳性的表达簇,其中包括 EPCAM,这一基因常用于识别 TNBC 患者的肿瘤细胞(图3c)。在四例肿瘤(TNBC1、TNBC2、TNBC4、TNBC5)中,上皮评分阳性的簇(c1–c2)与 CopyKAT 预测的非整倍体簇高度一致;然而,在 TNBC3 中,三个上皮基因评分阳性的簇中,仅有一个(c1)表现出全基因组CNA。将预测的非整倍体肿瘤细胞与 TNBC3 的上皮细胞表达簇(c1)比较,该研究估计 TNBC 样本中肿瘤细胞识别的准确率高达 98%。值得注意的是,在三例 TNBC(TNBC1、TNBC2、TNBC5)中,推断的拷贝数谱分布于两个肿瘤特异性表达簇,这些簇均为上皮基因评分阳性,提示肿瘤组织中存在多个非整倍体克隆。与 PDAC 和 ATC 肿瘤相比,TNBC 样本的肿瘤纯度普遍较高(34%–83%)(图3f)。
综合来看,这些数据表明,CopyKAT 能够利用scRNA-seq推断的非整倍体拷贝数谱,在多种实体瘤中准确区分肿瘤细胞与正常细胞(准确率 98%±3%),且无需依赖特定的基因表达标记。
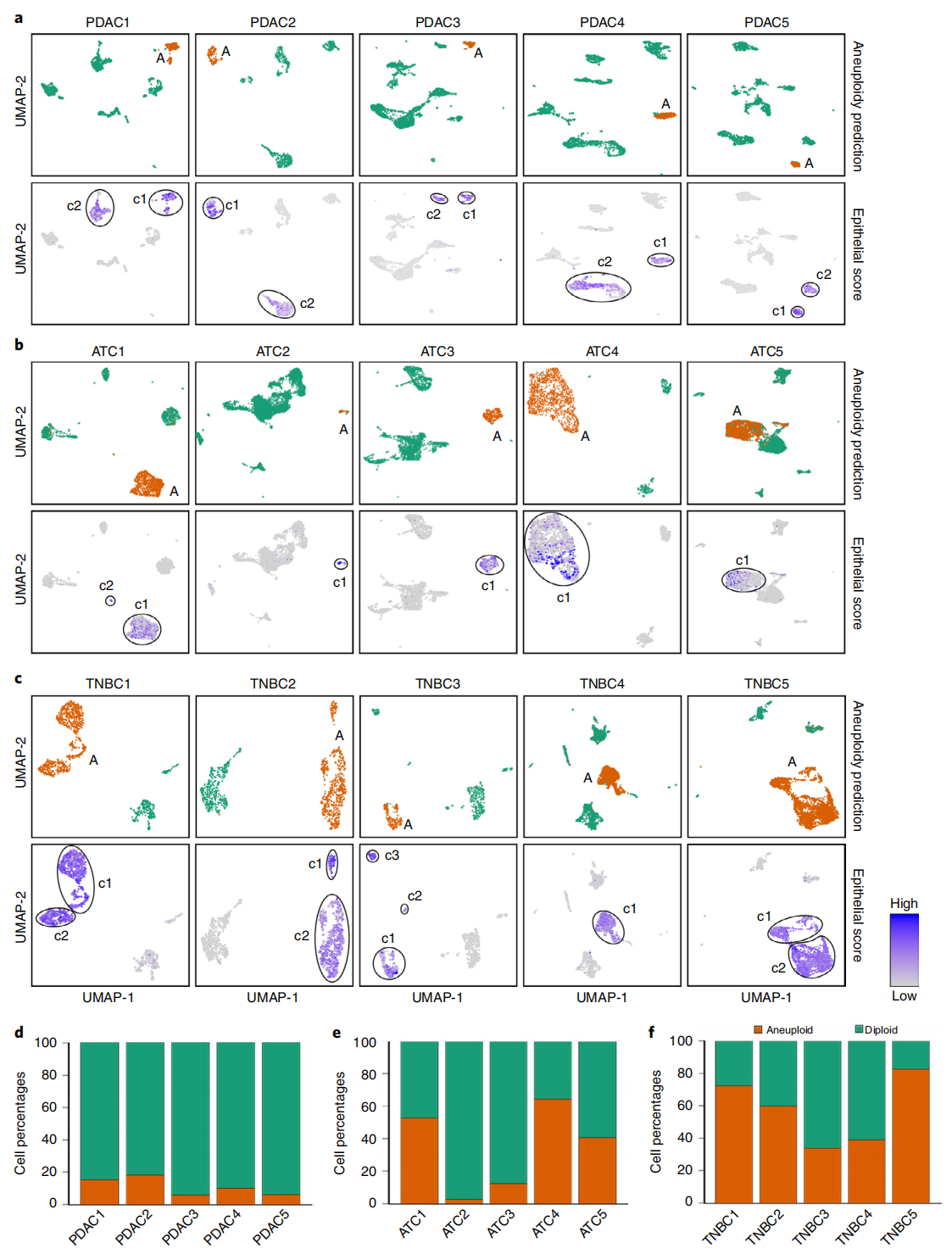
图3:人类肿瘤中癌细胞与正常细胞的分类
在其他单细胞 RNA 测序技术中的应用
在验证了 CopyKAT 对 3′scRNA-seq 数据的适用性后,研究进一步探讨了该方法在第一代 scRNA-seq 数据(SMART-seq2)以及 5′scRNA-seq 数据(10X Genomics)中应用的可行性。该研究对两例雌激素受体阳性浸润性导管癌(ER+ IDC)肿瘤(IDC1、IDC2)进行了 5′ scRNA-seq 测序,同时分析了先前研究中两例多形性胶质母细胞瘤(GBM;GBM1、GBM2)的 SMART-seq2 数据。在 IDC1 和 IDC2 中,共测序 7,780 个单细胞(5′scRNA-seq,10X Genomics)。CopyKAT分析在每例肿瘤中识别出两个簇,分别代表正常细胞(N)和肿瘤细胞(T),两例肿瘤均表现出染色体8p(MYC)的大幅扩增(图4a,c)。对scRNA-seq 表达数据进行聚类分析显示,两例肿瘤中推断的非整倍体肿瘤细胞与高上皮评分的簇高度共定位(图4b,d),验证了 CopyKAT 预测的准确性。与 TNBC 样本类似,两例 ER+ IDC 肿瘤的肿瘤细胞比例均较高(分别为 97% 和 87%)。
接下来,该研究分析了先前发表的两例多形性胶质母细胞瘤(GBM)患者的 scRNA-seq 数据,这些数据使用第一代全长 scRNA-seq方法生成,分别为 GBM1(MGH125)和 GBM2(MGH128)(GSE131928)。与10X Genomics的高通量3′或5′scRNA-seq 方法仅覆盖部分基因体不同,全长SMART-seq2 数据的测序读长能覆盖整个基因转录本。然而,SMART-seq2 的单细胞通量较低(每例分别为 332 和 184 个细胞),且缺少 UMI 条码,无法缓解扩增偏差。为进行 CopyKAT 分析,该研究使用了 TPM/10 矩阵表示标准化的基因表达计数数据。在两个样本中,观察到非整倍体肿瘤细胞簇与二倍体正常细胞簇明显分离(图4e,g)。CopyKAT 推断的非整倍体肿瘤细胞簇中 EGFR 表达水平较高(图4f,h),该基因是 GBM 患者公认的肿瘤细胞标记。
综上,这些数据表明 CopyKAT 可兼容多种 scRNA-seq 技术。
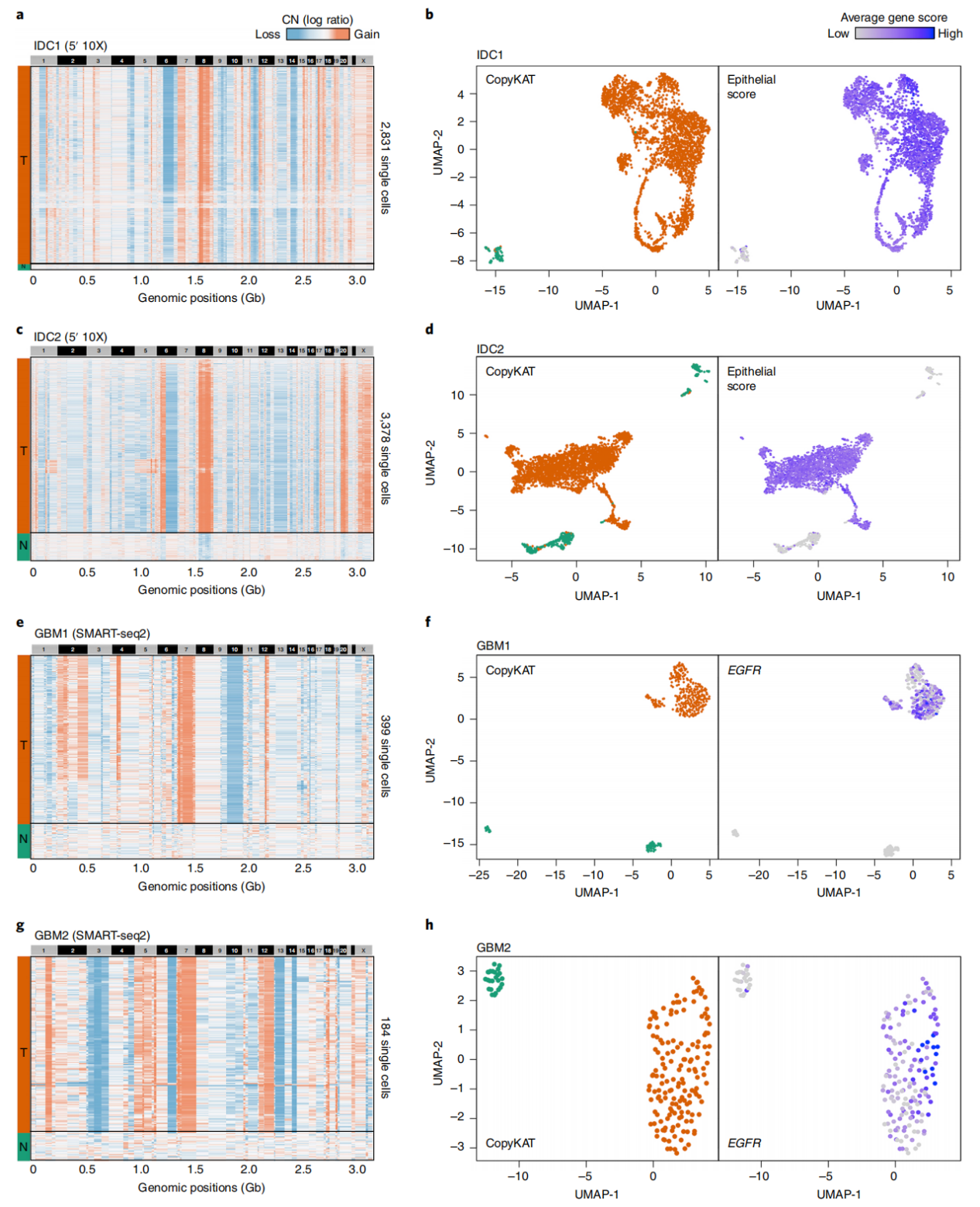
图4:使用不同 scRNA-seq 技术测序的肿瘤细胞与正常细胞分类
推断乳腺肿瘤的克隆亚结构
为解析克隆亚结构并将癌症基因型与表型关联,该研究将 CopyKAT 应用于三例三阴性乳腺癌(TNBC)患者的 scRNA-seq 数据(图5)。研究首先对推断的拷贝数谱进行聚类,识别出基于拷贝数差异的亚群,随后,从单细胞簇中计算共识拷贝数谱,以确定具有拷贝数差异的基因组区域。基于亚克隆的共识拷贝数谱,该研究进一步进行了差异表达(DE)分析和基因特征分析,以揭示亚克隆间的表型差异。
在一例 TNBC 样本(TNBC1)中,对797个单细胞非整倍体拷贝数谱进行聚类分析,识别出两个主要亚克隆(A、B),分别占肿瘤总量的 44% 和 28%(图5a,上方)。聚类热图显示所有肿瘤细胞共有的克隆性扩增(1q、6p、8q、10p、16p 和 18p)及克隆性缺失(1p、4q、5q、8p、10q、13 和 14)。这些基因组区域涵盖了TCGA数据库中记录的多个已知乳腺癌相关基因,如MDM4、PIK3CA、EGFR、MYC、GATA3、PTEN、CCND1、RB1等(图5a,上方)。共识拷贝数谱的聚类热图揭示了亚克隆 CNA 事件,包括亚克隆 A 的扩增(4p、7q、9p13.2–q22.2 和 17q)以及亚克隆 B 的扩增(3p26.3–p25.1、6q、7p、11q、Xp11.23 和 Xq),这些扩增在肿瘤中存在变异(图5a,下方)。差异表达分析(DE)共鉴定出两个亚克隆间329个差异基因(FDR调整P值 < 0.01,|log2(倍数变化)| ≥ 0.5),其中 47% 位于亚克隆 CNA 区域,包括已知癌基因,如位于 2q 染色体的 IDH1 在亚克隆 A 中过表达,以及位于 16q 的 CDH1 在亚克隆 B 中过表达(图5a,下方)。这两个非整倍体亚克隆在高维空间中对应不同的表达簇(图5d,上方)。单细胞基因集变异分析(GSVA)显示,亚克隆 A 相对于亚克隆 B 富集了若干癌症标志性通路,包括雄激素反应、上皮-间质转化(EMT)及其他癌症相关通路(图5d,下方)。
在另一例 TNBC 患者(TNBC2)中,对 620 个由 scRNA-seq 数据推断的单细胞非整倍体拷贝数谱进行聚类分析,识别出两个亚克隆,包括占肿瘤主体的主要亚克隆 A(53%)和占肿瘤小部分的次要亚克隆 B(7%)。聚类热图显示,所有肿瘤细胞共有的克隆性扩增区域包括 1、2p、6、8、9p 和 Xq,克隆性缺失区域包括 4q、5、9q、13、14、15、16q、20 和 Xp,这些区域涵盖了已知乳腺癌基因,如 MDM4、EGFR、MYC、CDKN2A、GATA3、PTEN、BRCA2、RB1、TP53 等(图5b,上方)。共识拷贝数谱比较进一步揭示了亚克隆特异性 CNA:亚克隆 A 特有的扩增为 16p13.3–p13.2,亚克隆 B 特有的扩增为 3q、12p13.1–q12 和 12q21.33–24.12。亚克隆 B 中出现的主要亚克隆事件为染色体 12p13.1–q12 的局部扩增,导致 KRAS 的过表达(图5b,下方)。次要亚克隆 B 的拷贝数谱在 scRNA-seq 高维分析中映射到一个独特区域(图5e,上方)。单细胞 GSVA 分析显示,主要亚克隆 A 中 WNT 和 Hedgehog 信号通路活性增强,而次要亚克隆 B 中多个癌症标志性通路上调,包括干扰素反应、TNF-α 信号、缺氧及其他通路(图5e,下方)。
在第三例 TNBC 患者(TNBC5)中,对 2,670 个由 scRNA-seq 数据推断的非整倍体单细胞拷贝数谱进行聚类分析,识别出两个亚克隆,包括占肿瘤主体的主要亚克隆 A(65%)和占肿瘤小部分的次要亚克隆 B(18%)。在所有肿瘤细胞中检测到的克隆性 CNA 包括 4q、7p 和 8q 的扩增,以及 1p、2q、9、10、11 和 17p 的缺失,这些区域涵盖多个乳腺癌基因,如 MDM4、PIK3CA、FGFR4、EGFR、MYC、TP53、CDKN2A 等(图5c,上方)。亚克隆共识拷贝数谱比较显示了若干亚克隆特异性 CNA,例如亚克隆 A 特有的 1p 和 14p 扩增,以及亚克隆 B 特有的 12p 和 12q 扩增(图5c,下方)。两个亚克隆的非整倍体拷贝数谱在高维表达空间中映射到不同区域,提示它们具有不同的转录程序(图5f,上方)。值得注意的是,次要亚克隆 B 中出现染色体 12p13.33–q12 的大幅扩增,导致 KRAS 表达升高,这与前一TNBC 样本(TNBC2)中发现的次要亚克隆B一致。单细胞 GSVA 分析显示,亚克隆 A 的癌症标志性通路表现为干扰素-α反应和脂肪酸代谢上调,而亚克隆 B 则表现出血管生成、缺氧及上皮-间质转化(EMT)等通路的增强(图5f,下方)。
综上结果表明,CopyKAT 能够从 scRNA-seq 数据中解析肿瘤的克隆拷贝数亚结构,并识别肿瘤内部存在的亚克隆差异,包括乳腺癌相关基因和癌症表型的异质性。
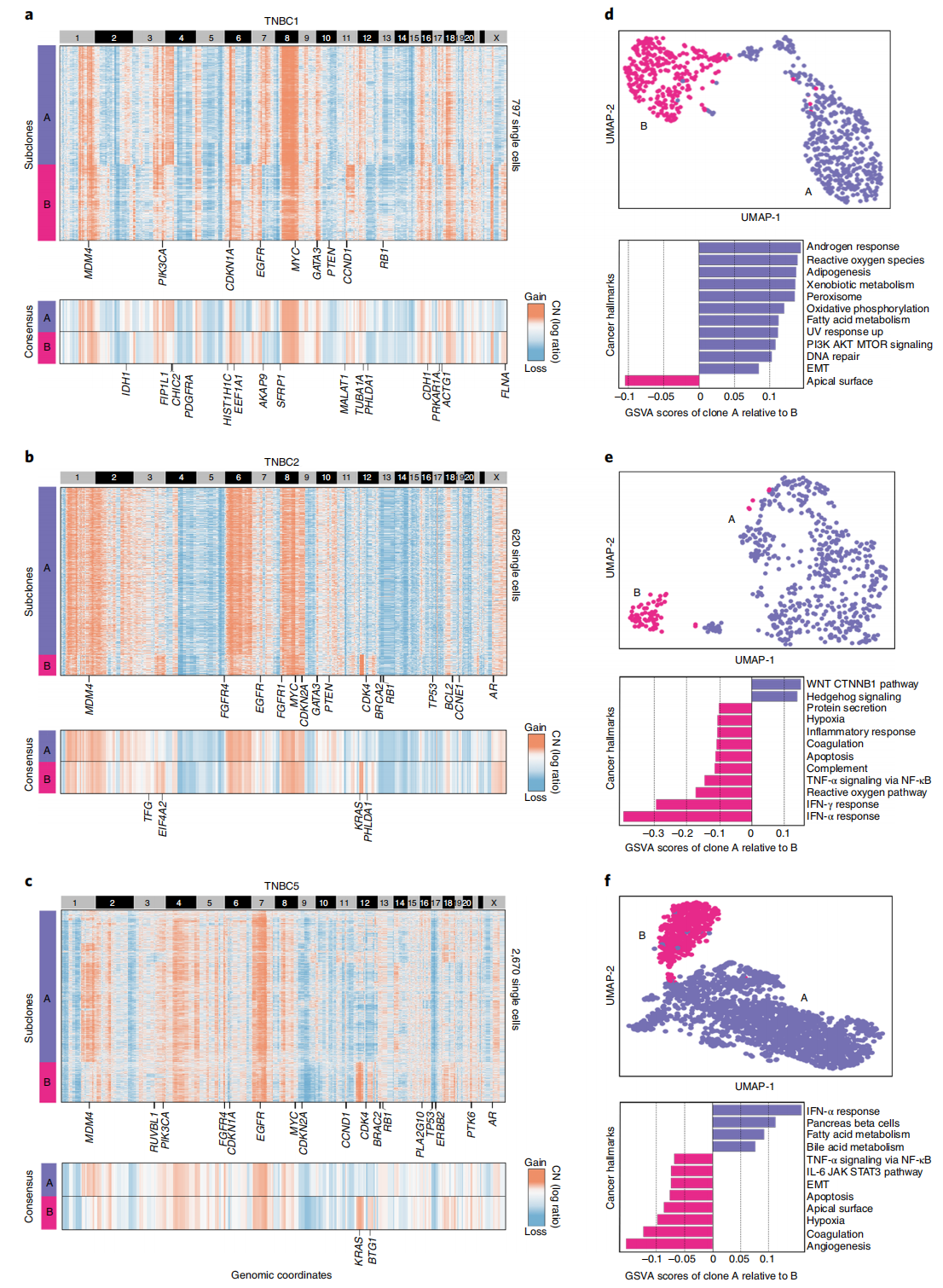
图5:三例三阴性乳腺癌的克隆亚结构
讨论:
该研究报道了一种整合贝叶斯分割方法,用于从高通量 scRNA-seq 数据中定量基因组拷贝数谱的新方法。CopyKAT 的一个主要应用是在无偏 scRNA-seq 数据中识别肿瘤细胞,这类数据通常不仅包含肿瘤细胞,还包含肿瘤微环境(TME)中的多种基质细胞和免疫细胞类型。正常上皮细胞往往是最难仅通过基因表达谱与恶性肿瘤细胞区分的细胞,因为它们可能表达许多与癌细胞相同的上皮标记。利用 CopyKAT,该研究利用了实体瘤中癌细胞的一个独特特性,即它们的基因组中通常存在非整倍体拷贝数事件,而大多数基质细胞和免疫细胞则具有二倍体拷贝数谱。该研究展示了,在包括胰腺导管腺癌(PDAC)、未分化甲状腺癌(ATC)、导管原位癌(DCIS)、三阴性乳腺癌(TNBC)、浸润性导管癌(IDC)以及多形性胶质母细胞瘤(GBM)等多种实体癌症类型中,从 scRNA-seq 数据分类非整倍体基因组是可行的,即便肿瘤纯度非常低(<15%)或非常高(>90%)。因此,该研究预计 CopyKAT 将成为在包含多种 TME 细胞类型混合的 scRNA-seq 实验中识别肿瘤细胞的有力工具。
CopyKAT 的另一个应用是基于拷贝数变异(CNA)差异解析实体肿瘤的克隆亚结构。该研究将 CopyKAT 应用于三例 TNBC 肿瘤,成功解析出每个肿瘤中两个主要亚群,这些亚群在特定的 CNA 事件上存在差异。此外,该研究表明,通过这些数据可以将亚克隆的基因型与其表型(转录程序)关联,从而理解基因组改变如何影响不同的癌症特性。该研究在 TNBC 中的分析显示,肿瘤内部的亚克隆在基因特征和信号通路上存在差异,包括上皮-间质转化(EMT)、DNA 修复、缺氧、凋亡和血管生成的变异。有趣的是,在两例 TNBC 肿瘤中,该研究还识别出罕见亚克隆(分别占 7% 和 18%),其在 12p 染色体上存在 KRAS 致癌基因扩增,并导致基因表达上调。如果在未来更大规模的 TNBC 患者队列中发现这些罕见 KRAS 亚克隆是常见亚群,它们可能成为诊断或治疗的潜在靶点。
此前已有两种方法用于估算拷贝数变异(CNA)。inferCNV 在剔除高表达和低表达基因后,通过平均滑动窗口计算基因表达;然而,inferCNV 在精确解析染色体断点方面能力有限。另一种方法 HoneyBadger10 旨在通过联合分析单细胞聚类中多个变异位点的等位基因不平衡来预测 scRNA-seq 数据的拷贝数变异,但其高度依赖于获得基因体的全覆盖数据。因此,这些方法的一个局限性是,它们不适用于目前广泛使用的 3′和 5′scRNA-seq 方法(如 10X Genomics、Drop-Seq、InDrop),而是针对第一代 scRNA-seq 方法开发的,例如 Fluidigm35 和 SMART-seq。相比之下,CopyKAT 兼容高通量 scRNA-seq 方法,可对数千个并行测序的细胞生成稀疏数据(例如每个细胞 10 万条测序读数),同时也兼容第一代 scRNA-seq 方法的数据。
CopyKAT的一个显著局限性是,并非所有癌症类型都具有可用于区分正常细胞与肿瘤细胞的非整倍体拷贝数事件。特别是,小儿癌症和血液系统癌症(例如急性髓系白血病 AML 和慢性淋巴细胞白血病 CLL)已知具有较少的拷贝数变异,因此可能不适合使用 CopyKAT 进行分析。另一个局限性是,CopyKAT主要依赖于基于基因组中测序深度变化来检测 CNA 事件,无法用于检测其他影响基因组多样性的基因组事件,包括染色体结构重排、插入缺失(indels)以及体细胞突变。此外,由于 3′scRNA-seq 数据的技术变异性,CopyKAT 无法提供单个具有独特基因型细胞的可靠拷贝数信息。这使得 CopyKAT 更适合用于分析那些大量细胞扩增且共享相似基因型的肿瘤亚克隆,而不适合用于分析正在复制的细胞或极其稀少的亚群。该研究注意到的一个潜在问题是,当 scRNA-seq 数据集中不存在任何肿瘤细胞时,CopyKAT 可能会尝试在基因表达水平最高的聚类中错误地检测 CNA 事件。在这种情况下,推断出的 CNA 事件将与这些癌症的已知细胞遗传事件不一致,因此可以被忽略。
CopyKAT 提供了一种强大的自动化工具,用于对高通量 scRNA-seq 方法分析的实体肿瘤中的肿瘤细胞和正常细胞进行分类,并解析克隆亚结构。该研究预计,这一工具不仅可应用于本研究分析的癌症类型,还可广泛应用于多种实体肿瘤的研究。通过提供肿瘤细胞信号,这些研究将大大改善对肿瘤细胞恶性基因表达程序的理解,而以往的整体 RNA-seq 方法由于肿瘤细胞与基质细胞及免疫细胞的混合,往往难以准确评估癌症表型。此外,这些研究将有助于揭示染色体改变如何通过基因剂量效应重新编程人类肿瘤在疾病进展中的癌症表型,从而获得新的生物学见解。

汇报人:伍施宇
审核:夏晓旭、冯兰、任建君
