


原创 黄石 华西医院耳鼻喉科
阅读最新文献,紧跟前沿进展,这是一名研究者必须具备的习惯和要求。我们华西医院耳鼻咽喉头颈外科的硕士、博士研究生和博士后们自2019年以来,每周开展一次文献泛读和文献精读分享会,至今已累计开展了200多次。2023年9月13日开始,本科室陆续将其进行整理,同步推出在线前沿速递和文献解读板块。通过这种学习和分享的方式,使汇报者和大家都能对近期权威期刊发表的高质量研究有所了解,同时也是学习其他优秀研究者思路、方法和理论的良好手段。希望通过这种形式,把科内的分享扩大到所有的读者,一起学习,共同进步!
华西医院耳鼻咽喉头颈外科

精读分享│【Nature Human Behaviour】:跨血统全基因组关联研究及系统层面综合分析识别抑郁症新风险基因和治疗靶点
英文题目:Cross-ancestry genome-wide association study and systems-level integrative analyses implicate new risk genes and therapeutic targets for depression
中文题目:跨血统全基因组关联研究及系统层面综合分析识别抑郁症新风险基因和治疗靶点
期刊:Nature Human Behaviour(IF: 21.4)
单位:东南大学,中国科学院,昆明医科大学,郑州大学,QIMR Berghofer医学研究所,武汉大学,浙江大学医学院等
发表时间:2025年2月



摘要:
解析抑郁症的遗传架构对于揭示相关的病理生理过程和开发新型治疗方法至关重要。该研究开展了一项跨祖先人群的全基因组荟萃分析(GWAS-META),纳入了416,437例抑郁症病例和1,308,758名对照,共鉴定出287个风险位点,其中49个为新发现的位点。在变异水平上,精细定位分析优先筛选了潜在的因果变异,而功能基因组学分析进一步鉴定了能够调控转录因子结合的功能性变异。实验验证表明,80%的功能性变异具有调控活性,同时,基因表达数量性状位点(eQTL)分析揭示了这些优先筛选的风险变异所调控的潜在靶基因。在基因水平上,采用了转录组(TWAS)和蛋白组(PWAS)广泛关联分析、共定位分析及孟德尔随机化分析(SMR),筛选出潜在的因果基因及药物靶点。基因优先排序分析筛选出TMEM106B、CTNND1、AREL1等可能的致病基因。通路分析结果表明,抑郁症风险基因显著富集于突触相关信号通路。此外,Tmem106b基因敲低小鼠出现抑郁样行为,进一步支持了Tmem106b在抑郁症发生中的作用。该研究鉴定了新的抑郁症风险位点、可能的因果变异及基因,为揭示抑郁症的遗传架构和寻找潜在治疗靶点提供了重要线索。
前言:
抑郁症是全球最常见的精神障碍之一,也是导致残疾的主要原因。尽管近年来全基因组关联研究(GWAS)已报道了多个抑郁症的风险位点,但其遗传基础仍未被完全阐明。此外,大多数抑郁症GWAS研究主要针对欧洲人群进行,这可能导致部分关键的遗传因素被忽视。更重要的是,对于大多数已报道的风险位点,其具体的因果变异及相关基因仍不明确,限制了遗传研究成果向临床应用和治疗的转化。因此,鉴定新的遗传风险位点,并对已识别的风险变异及基因进行功能解析,将有助于深入理解抑郁症的病理生理机制,并发现潜在的治疗靶点。
该研究首先开展了一项大规模的跨血统人群荟萃分析(包括416,437例病例与1,308,758名对照)。基于荟萃分析的结果,进一步进行了系统的优先筛选及整合分析,以鉴定潜在的因果变异和基因。研究筛选出可能的因果变异,并验证了所鉴定的功能性变异在转录调控方面的作用。此外,还筛选出多个可能的因果基因,包括TMEM106B、CTNND1、EPHB2等。最后,小鼠模型实验显示,Tmem106b基因敲低可导致抑郁样行为,进一步支持TMEM106B作为抑郁症风险基因的作用。该研究鉴定了新的抑郁症风险位点、可能的因果变异及相关基因,为解析抑郁症的遗传架构及发现潜在治疗靶点提供了重要参考。
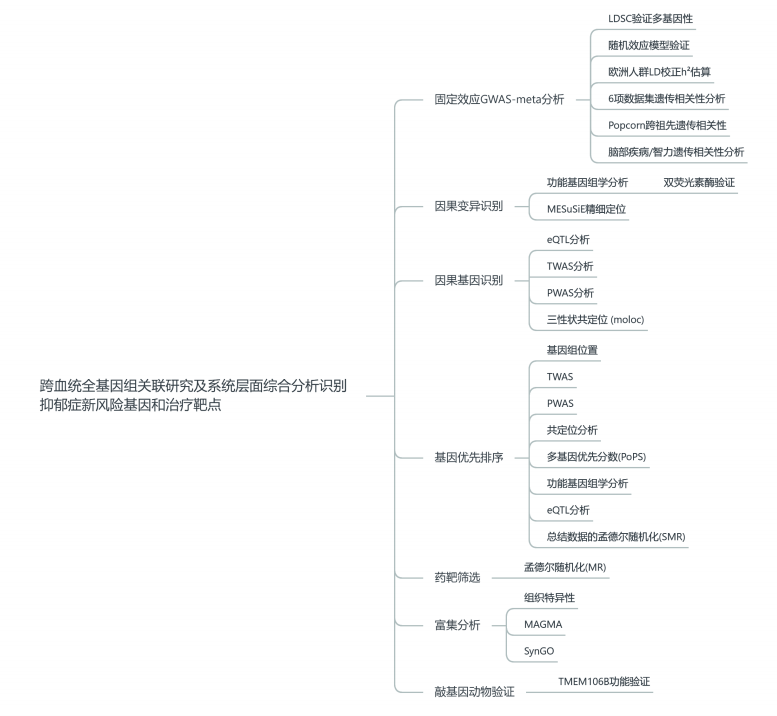
思维导图
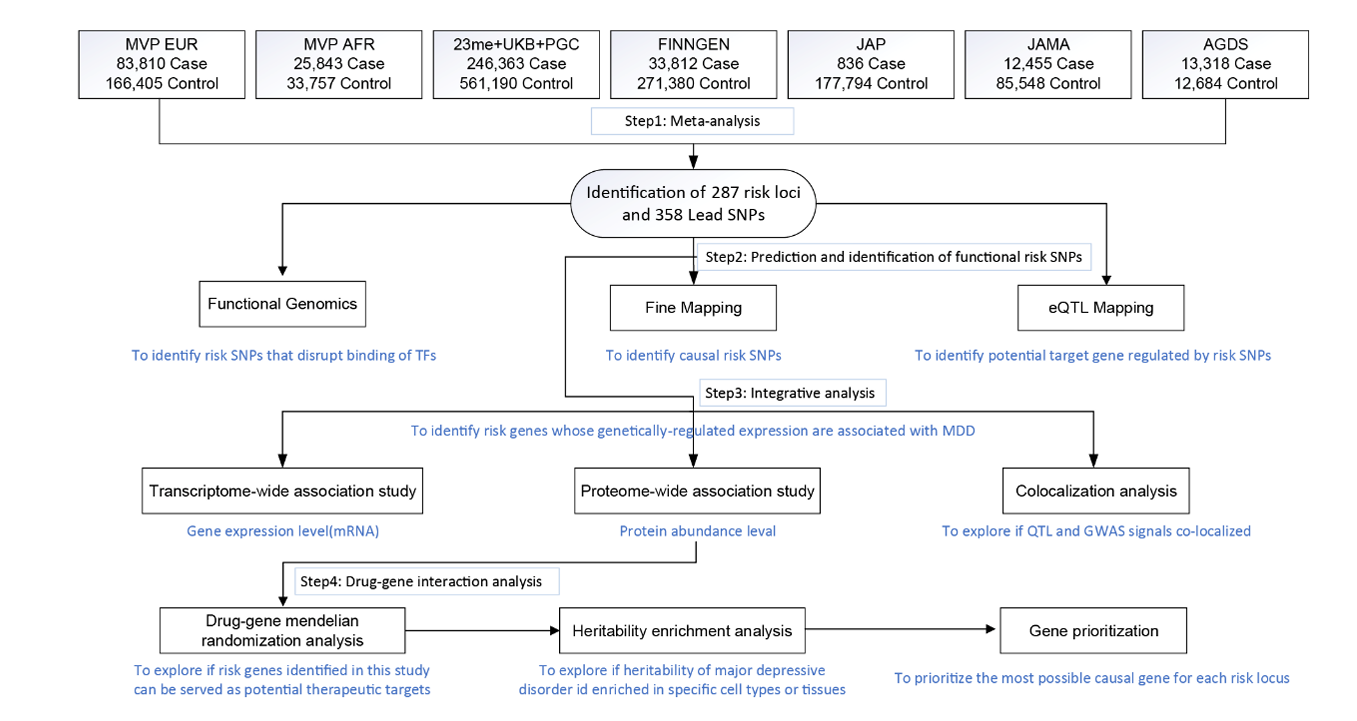
研究路线图
结果
1、跨祖先人群的荟萃分析鉴定出287个抑郁症风险位点
该研究整合了六项既往研究的数据,进行抑郁症的全基因组关联分析。研究人群包括来自美国的 Million Veteran Project(MVP)欧洲裔和非洲裔队列、芬兰的FinnGen研究、由Howard等人报道的UK Biobank(UKB)+精神基因组学联盟(PGC)+ 23andMe数据、澳大利亚抑郁症遗传学研究(AGDS)、Giannakopoulou 等人报道的东亚多个队列,以及Sakaue等人报道的日本生物银行(BioBank Japan)。研究采用固定效应模型进行荟萃分析(共纳入416,437例病例和1,308,758名对照),共鉴定出287个独立的基因组风险位点(P < 5×10-8),其中49个为新发现的风险位点。
在鉴定的风险位点中,最显著的单核苷酸多态性(SNP)rs753111,位于 NEGR1 基因(1p31.1)下游约89kb处,而第二显著的SNP rs1021363位于SORCS3 基因内含子2区域。连锁不平衡评分回归(LDSC分析表明,研究结果主要受多基因作用影响,而非混杂因素(基因组控制lambda值1.54,截距1.04 ±0.01,比率 0.03±0.008)。
此外,为评估等位基因效应在跨血统人群荟萃分析中的变异性,该研究针对具有异质性的变异(P_Cochran’s Q < 0.05)进一步采用随机效应模型进行荟萃分析。结果显示,在固定效应模型鉴定的287个风险位点中,272个在随机效应模型中也得到了支持,表明这些关联结果具有较强的稳健性。该研究的发现进一步拓展了抑郁症的遗传风险位点。
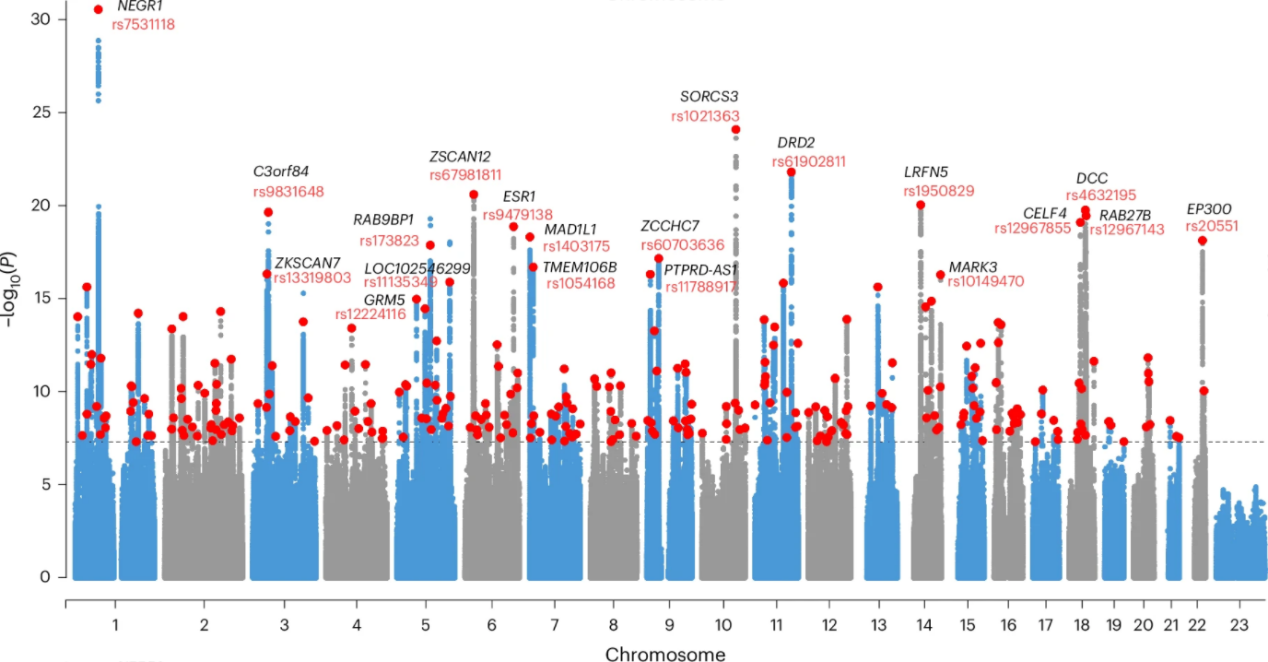
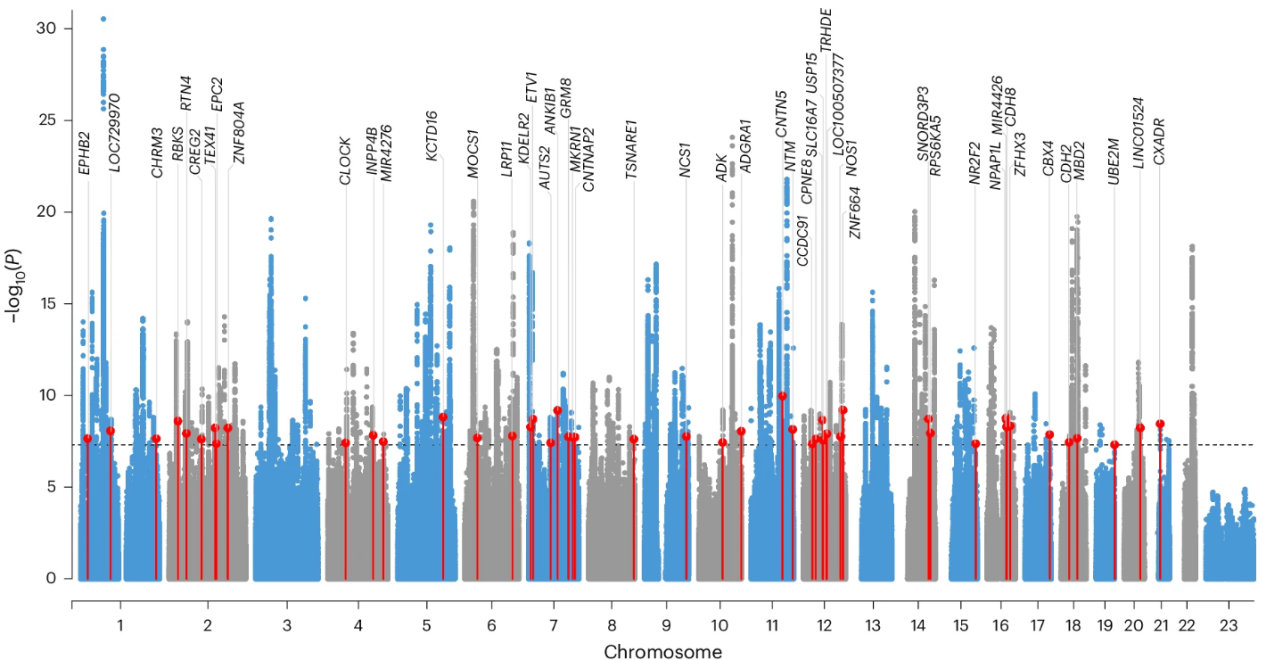
2、遗传力和遗传相关性
考虑到在使用跨血统人群样本估计单核苷酸多态性(SNP)遗传力(h²)时可能存在的连锁不平衡(LD)参考面板不匹配效应,研究使用了来自欧洲血统人群的全基因组关联研究(GWAS)荟萃分析(377,303例病例和1,011,659名对照)来估计遗传力。假设终生风险为0.15时,基于责任尺度遗传力(liability scale h²)估计值为0.073(通过LDSC估算,标准误差为0.003)和0.095(使用LDAK软件包中的SumHer方法结合基线-LD模型计算,标准误差为0.002)。这些值与先前报道的结果一致(遗传力在liability scale上的范围为0.080–0.090)。
研究进一步计算了六项抑郁症研究之间的遗传相关性(MVP、FinnGen、AGDS、23andMe–UKB–PGC、Howard等人和Sakaue等人)。考虑到低遗传力性状的遗传相关性估计通常伴随较大的混杂,因此其结果可能缺乏可靠性,不宜作为有效结论,研究将重点放在了遗传相关性Z分数大于4的GWAS数据集(包括23andMe–UKB–PGC、AGDS、FinnGen、MVP欧洲祖先人群(EUR)),并发现不同抑郁症研究之间的遗传相关性高度显著。鉴于不同祖先人群中的遗传结构和LD结构差异,研究进一步使用了Popcorn工具,该工具能够计算跨血统人群的遗传相关性,以估算东亚人群(EAS)和欧洲人群(EUR) GWAS数据集之间的遗传相关性(仅包含SNP遗传相关性Z分数大于2的数据集)。Popcorn的结果也表明包含的数据集之间存在较高的遗传相关性。同时,基于LD截距(1.04,标准误差为0.010)和衰减比(0.03,标准误差为0.008)的证据也表明,该研究的荟萃分析结果中没有显著的膨胀或混杂效应。
最后,该研究探讨了抑郁症(即来自欧洲祖先人群的荟萃分析结果,包括MVP-EUR、23andMe–UKB–PGC、AGDS和FinnGen数据集)与其他脑部疾病和智力之间的遗传相关性。与抑郁症表现出最显著遗传相关性的前三种疾病是焦虑症、创伤后应激障碍和神经质。大多数分析的性状与抑郁症呈正相关,然而,智力则显示出负相关。
3、功能基因组学识别了潜在的因果变异
识别功能性(或潜在的因果)变异对于后续机制研究和功能表征至关重要。为了从已识别的风险位点中找出功能性变异,研究进行了功能基因组分析,如前所述。研究识别了64个影响转录因子(TFs)结合的功能性单核苷酸多态性(SNPs)(即TF结合影响SNPs)。其中,37个影响CCCTC结合因子(CTCF)的结合,10个影响RE-1沉默转录因子(REST)的结合,6个SNP同时干扰多个转录因子。约40%的TF结合影响SNP位于内含子区域,表明内含子变异在抑郁症中的重要作用。值得注意的是,34个干扰TF结合的SNP与抑郁症显著相关,其中影响CTCF结合的SNP rs7531118(位于NEGR1基因下游)显示出最显著的关联。研究结果揭示了来自已报告的风险位点的功能性(或潜在因果)变异,并表明影响转录因子结合是这些功能性SNP在抑郁症中发挥生物学作用的主要方式。
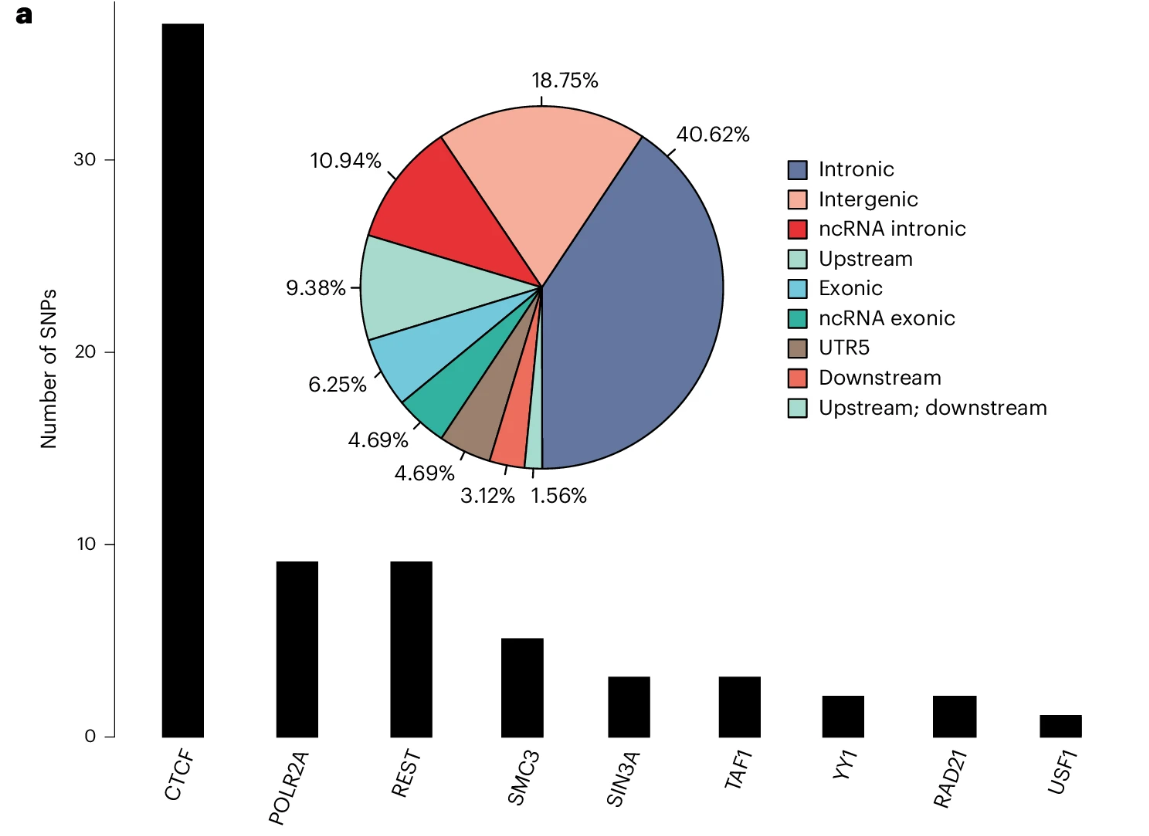
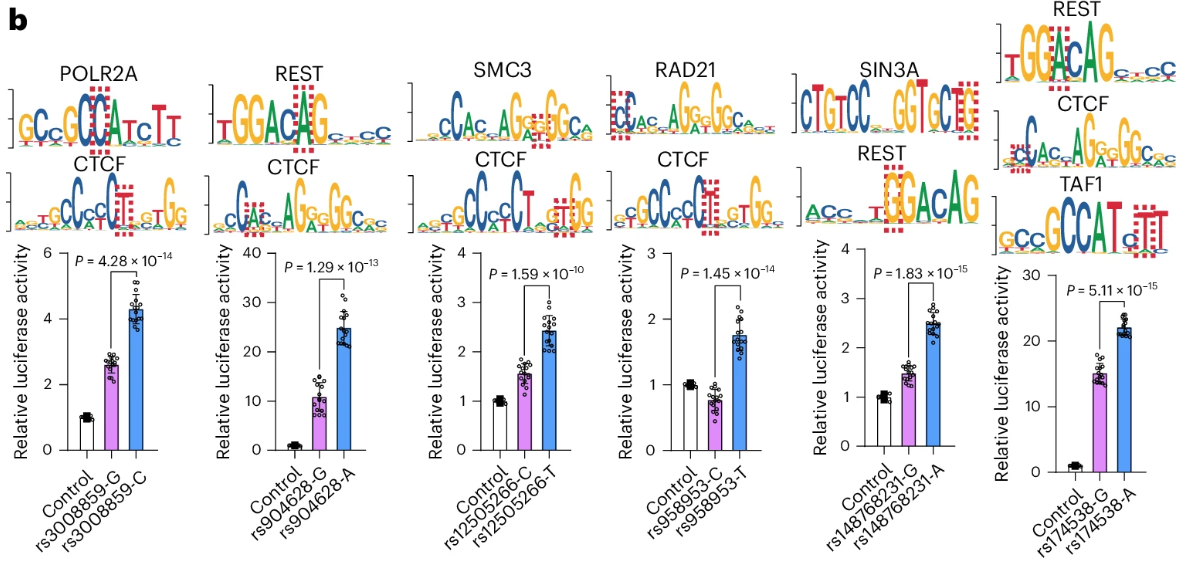
4、转录因子结合影响SNP的调控效应
为了验证已识别的转录因子结合影响SNP的调控效应,研究对所有转录因子结合影响SNP进行了双荧光素酶报告基因检测。报告基因检测结果表明,63个SNP中有51个显示出调控效应(其中1个SNP因其复杂的基因组序列未能成功构建载体),即这些51个SNP的不同等位基因显著影响了荧光素酶活性(P < 0.05)。这些结果提供了实验证据,支持大多数识别出的影响TF结合的SNP是调控变异。考虑到转录因子与调控序列的相互作用在基因表达调控中起着关键作用,这些发现也表明,这些功能性SNP可能通过调控基因表达来增加抑郁症的风险。这些结果为抑郁症风险变异的调控机制提供了重要的见解。
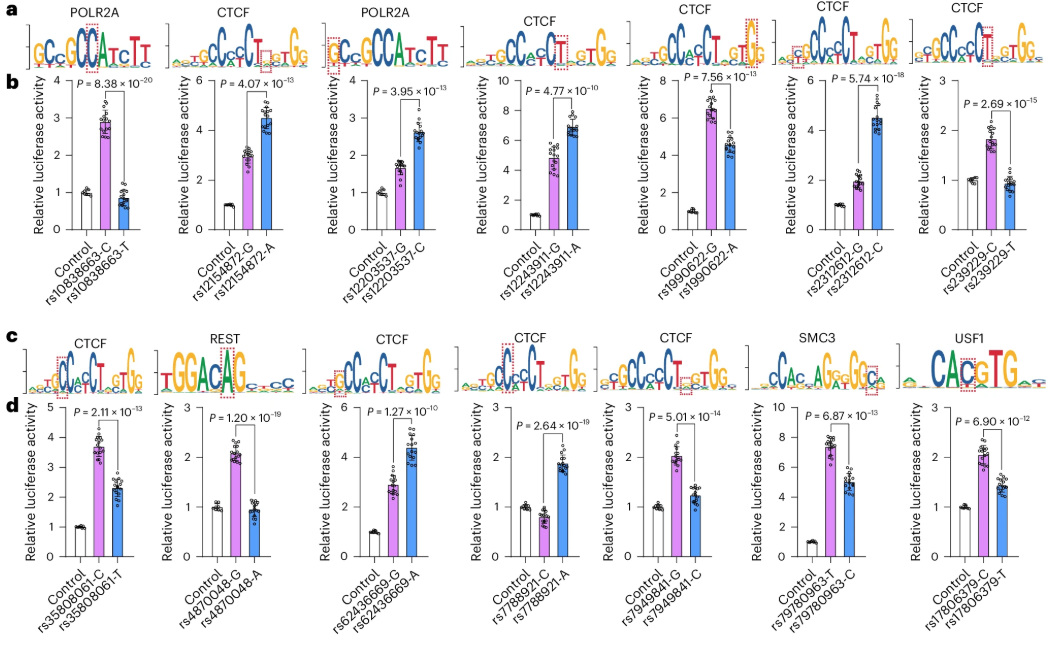
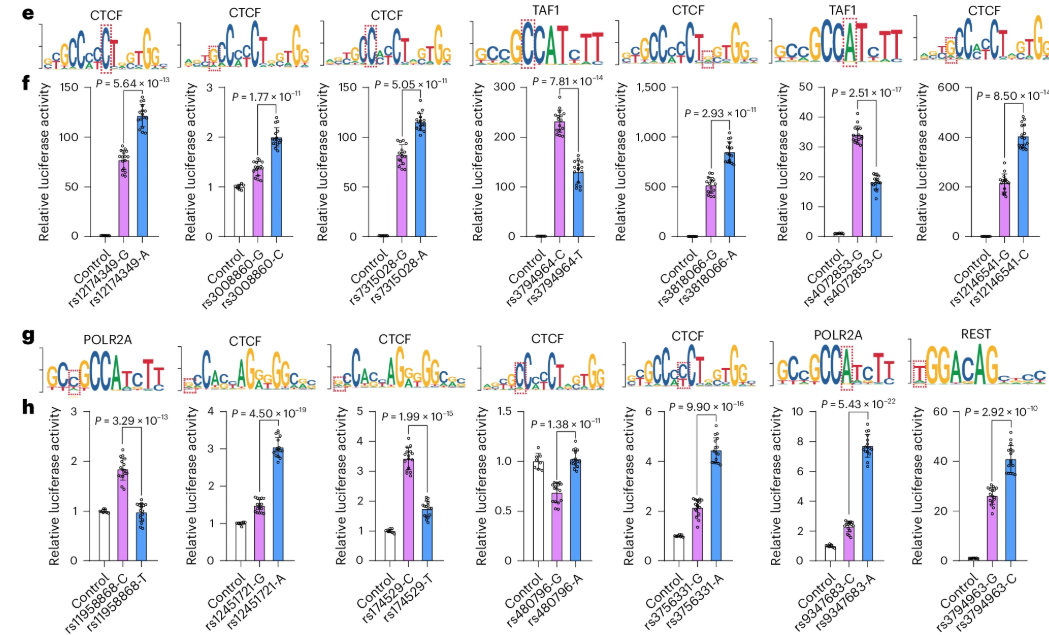
5、精细定位优先筛选潜在因果变异
为了从已识别的风险位点中筛选潜在的因果变异,研究采用了单效应模型精细定位方法MESuSiE,该方法利用多血统群体GWAS研究的关联信息进行精细定位。考虑到MESuSiE旨在识别两个血统人群之间共享的一组候选SNP,而本研究中包括的非洲血统人群并未识别出任何显著的风险SNP(GWAS P < 5×10−8),因此,研究仅使用亚洲和欧洲血统群体的GWAS数据进行精细定位。MESuSiE优先筛选出了122个高置信度的潜在因果变异(在亚洲或欧洲群体中的MESuSiE后验包含概率(PIP)>0.5),涉及208个风险位点。值得注意的是,MESuSiE还优先筛选出十个在亚洲和欧洲血统群体中共享的潜在因果SNP,这表明这些SNP可能在抑郁症中具有血统共享的因果效应。
6、风险变异的潜在靶基因
大多数已识别的抑郁症风险变异位于非编码区域,这表明基因表达的调控可能是风险变异与相关表型之间的重要潜在通路。为了识别这些风险变异的潜在靶基因,研究使用了BrainMeta v2数据集,该数据集包含来自2,865个人类大脑转录组的表达数量性状位点(eQTL)数据,以识别风险变异与人类大脑中基因表达之间的关联。研究使用了先导SNP、通过功能基因组学鉴定的TF结合影响SNP和通过MESuSiE优先筛选的因果候选SNP进行eQTL分析。对于先导SNP,161个SNP在经过Bonferroni修正后显示出显著关联(P < 3.90×10−5)。接下来,研究考察了TF结合影响SNP与基因表达之间的关联。在64个TF结合影响SNP中,50个与人类大脑中的基因表达显著关联(Bonferroni修正P < 2.40×10−5)。最后,研究发现36个通过MESuSiE优先筛选的SNP与人类大脑中的基因表达显著关联。综上所述,这些结果识别了由主要和优先筛选的功能性风险SNP调控的潜在靶基因,表明这些功能性变异通过调控这些靶基因的表达来增加抑郁症的风险。
7、TWAS识别了抑郁症风险基因
考虑到转录组广泛关联研究(TWAS)首先通过使用相对较小的参考面板(即TWAS依赖于特定族裔LD参考面板)构建SNP-基因表达权重,且eQTL数据来自欧洲祖先人群,研究整合了来自欧洲祖先人群的抑郁症GWAS结果和来自PsychENCODE(欧洲祖先人群)的脑部eQTL数据,进行了TWAS。使用FUSION工具识别了那些基因表达水平受到遗传调控并与抑郁症相关的基因。TWAS共识别出179个与抑郁症相关的基因,其遗传调控的表达水平与抑郁症显著相关。转录组广泛显著的基因包括RPL31P12、ZSCAN12P1、AREL1、HIST1H4L、RP11-73M18.6、TMEM106B等。值得注意的是,TMEM106B(位于主要SNP rs1054168附近)是TWAS中的一项重要关联。此外,功能基因组学还揭示了位于TMEM106B下游的功能性SNP rs1990622与抑郁症的强关联。与功能基因组学和eQTL分析一致,报告基因检测验证了rs1990622的调控效应,结果表明rs1990622的G等位基因与A等位基因相比显著提高了荧光素酶活性。这些发现不仅识别了与抑郁症相关的基因,还优先筛选了rs1990622作为一个功能性风险变异,表明其通过调控TMEM106B的表达影响抑郁症的风险。
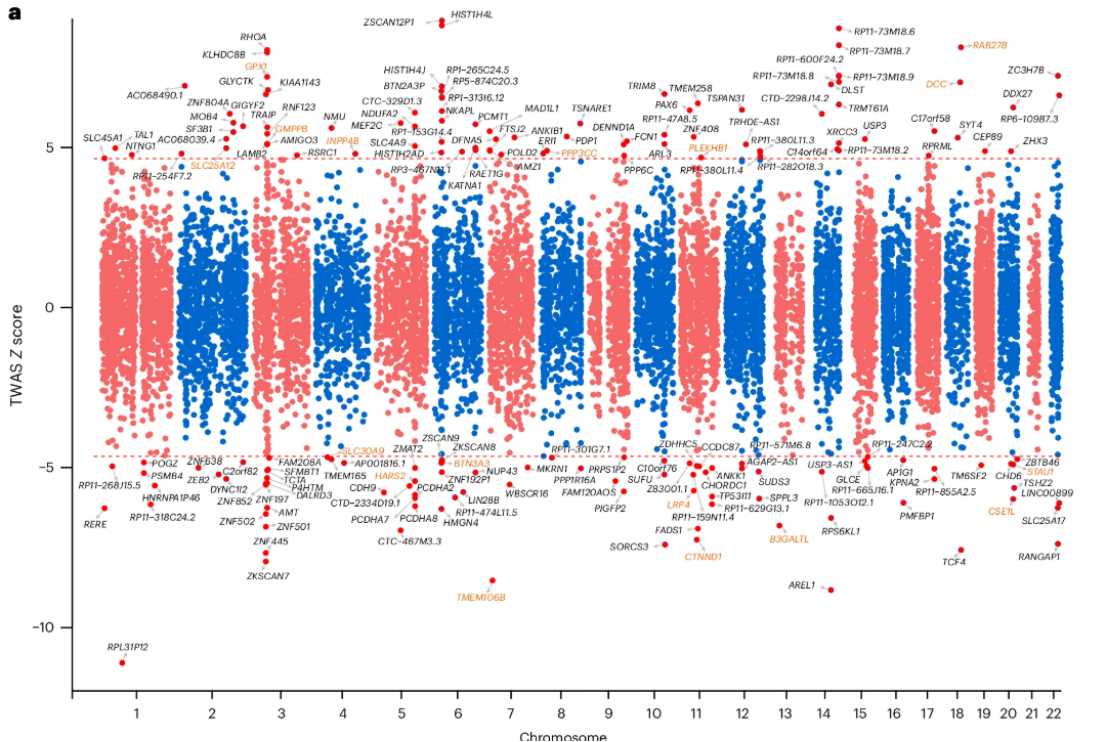
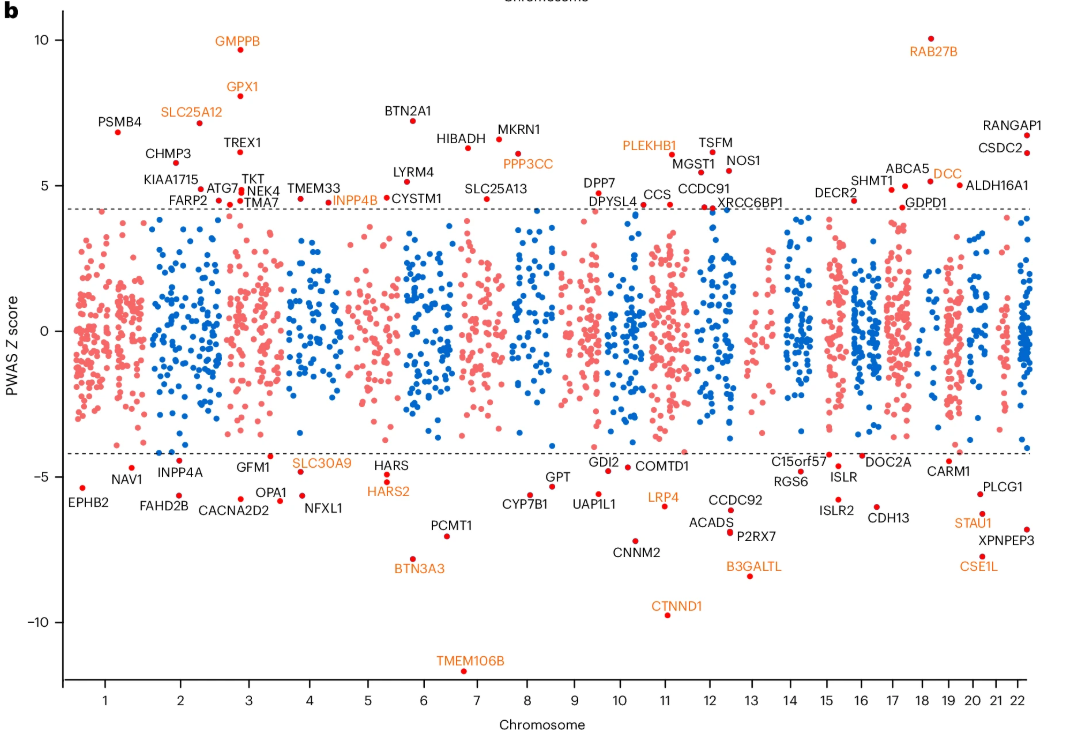
8、PWAS识别了与抑郁症相关的蛋白质
与TWAS分析类似,研究通过整合来自欧洲祖先人群GWAS数据集的抑郁症元分析结果以及来自两个人脑蛋白质定量性状数据集(均来自欧洲人群,分别是宗教订单研究/记忆与衰老项目(ROSMAP)和Banner数据集)的数据,进行了蛋白质组广泛关联研究(PWAS)。为了最大化PWAS分析的效能,研究进一步进行了元分析,将这两个数据集的PWAS结果进行整合。使用来自元分析的蛋白质定量性状位点(pQTL)数据,PWAS识别出了76个与抑郁症相关的蛋白质,其遗传调控的丰度与抑郁症显著相关。值得注意的是,21个蛋白质在转录组水平上也显示出了显著的关联,其中17个具有相同的效应方向,包括TMEM106B、RAB27B、CTNND1、GPX1、DCC、B3GALTL、CSE1L、LRP4、HARS2、GMPPB、SLC25A12、STAU1、PPP3CC、BTN3A3、INPP4B、PLEKHB1和SLC30A9,强烈暗示这些基因是潜在的抑郁症风险基因。有趣的是,Deng等人进行的孟德尔随机化研究也发现,RAB27B、GMPPB和TMEM106B的表达丰度在蛋白质和mRNA水平上都与抑郁症风险相关,提示这些基因可能在抑郁症中具有潜在的因果效应。考虑到抑郁症与焦虑障碍之间显著的遗传相关性,研究还比较了本研究中识别的风险蛋白与之前在焦虑障碍的PWAS研究中识别的蛋白质。包括TMEM106B、RAB27B和CTNND1在内的三种蛋白质与抑郁症和焦虑障碍均有显著关联。这些数据强烈提示TMEM106B、CTNND1、GMPPB和RAB27B是抑郁症的潜在候选基因。
9、共定位分析
为了探究GWAS和QTL信号是否由相同的变异驱动,研究进行了共定位分析。使用来自PsychENCODE的eQTL和GWAS信号进行的共定位分析显示,126个基因存在共享变异(PP4 > 0.70),包括RPL31P12、AREL1、TMEM106B、RAB27B、KLHDC8B、ZKSCAN7等。当将共定位分析限制在转录组广泛显著基因(Bonferroni校正后的P值阈值为3.39×10−6)时,65个基因显示出共定位信号。
ROSMAP pQTL和GWAS信号的共定位分析识别出了34个候选蛋白质,其中包括TMEM106B(PP4 0.995)、B3GALTL(PP4 0.987)、CNNM2(PP4 0.987)、RAB27B(PP4 0.985)等。Banner pQTL和GWAS信号的共定位分析识别出了24个蛋白质。值得注意的是,十个基因在三组QTL数据集的共定位分析中得到了支持,包括TMEM106B、SLC25A12、RAB27B、PAMB4等,这些基因提示它们是抑郁症的候选基因。
考虑到标准的共定位分析一次仅能推断两个性状(例如GWAS和eQTL)之间的共定位关系,研究进一步使用moloc包进行三性状(即GWAS、eQTL和pQTL)的共定位分析。值得注意的是,moloc分析中支持的六个基因包括NCOA3、SULF2、RNU7-173P、SRMP1、RNA5SP486和RNU7-92P,这些基因可能是抑郁症的高可信度候选基因。
10、潜在治疗靶点的识别
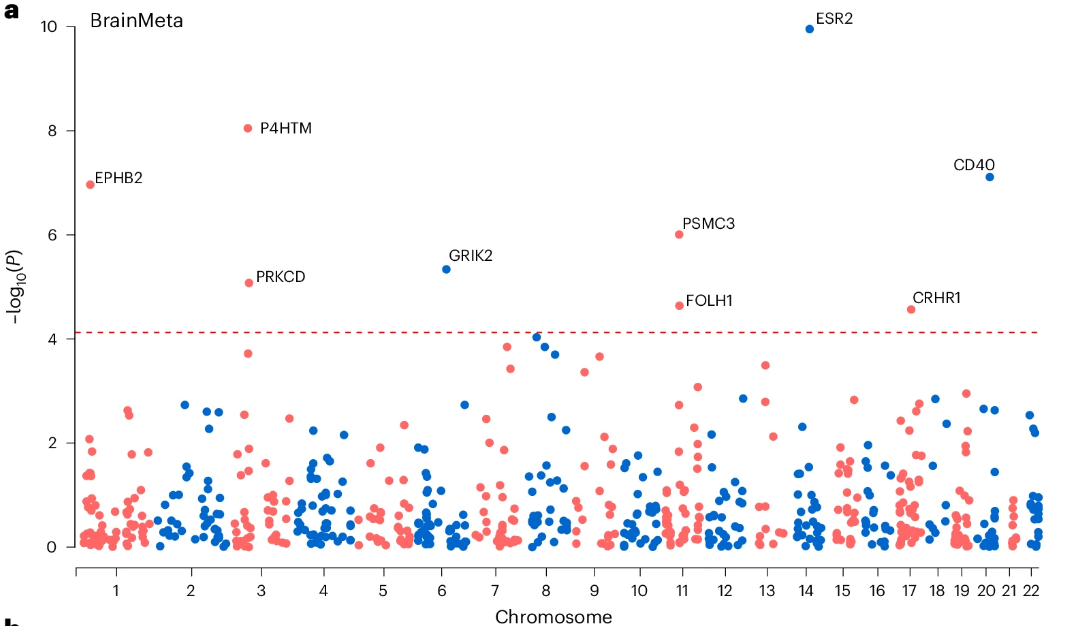
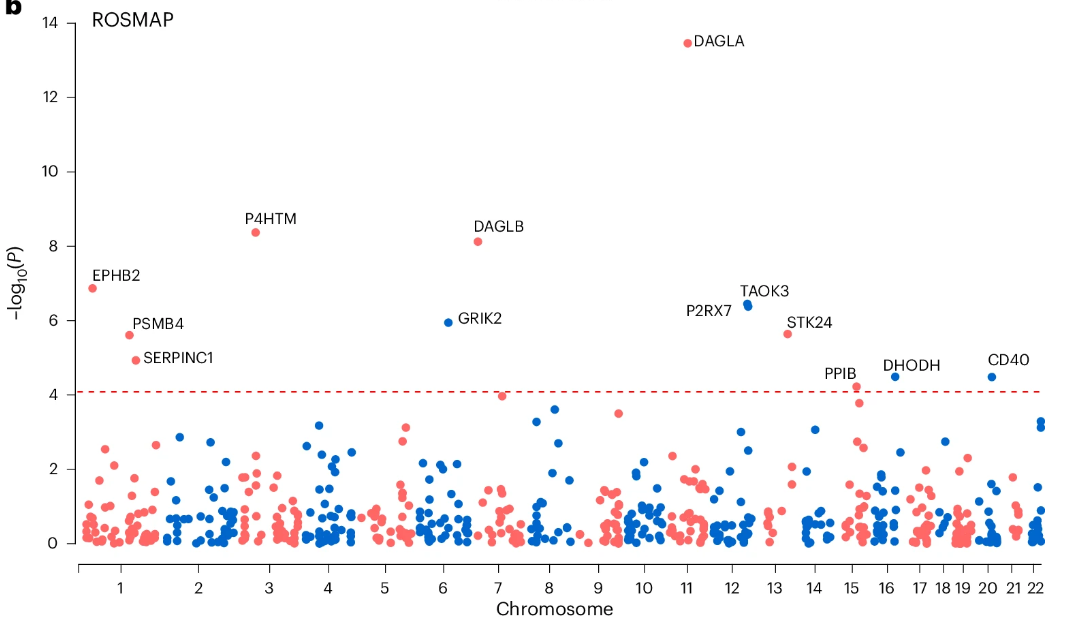
为了识别新的药物靶点并寻找潜在的药物再利用机会,该研究使用与1,263个可操作药物蛋白(已批准或临床阶段药物的治疗靶点)相关的基因变异进行了孟德尔随机化(MR)分析,这些靶点由Gaziano等人整理。
使用来自BrainMeta v2数据集的eQTL和GWAS信号的MR分析识别了九个有前景的可操作药物靶点(Bonferroni校正后的P值阈值<7.50 ×10−5),包括ESR2、P4HTM、CD40、EPHB2、PSMC3、GRIK2、PRKCD、FOLH1和CRHR1等。使用ROSMAP数据集的pQTL和GWAS关联的MR分析识别了13个有前景的候选蛋白质,包括DAGLA、P4HTM、DAGLB、EPHB2、TAOK3等。使用Banner数据集的pQTL和GWAS关联的MR分析识别了八个蛋白质。值得注意的是,DAGLA、P2RX7和PSMB4在两个pQTL数据集的MR分析中得到了支持。P4HTM、CD40、EPHB2和GRIK2在mRNA和蛋白质水平都显示出MR显著性,这表明这些基因是抑郁症的潜在药物靶点。
为了从MR结果中识别潜在的治疗药物,该研究根据MR结果的效应方向为每个可能的致病基因或蛋白质突出显示了潜在的治疗剂。例如,如果MR beta > 0,则列出了针对该基因或蛋白的抑制剂/拮抗剂,而如果MR beta < 0,则列出了激动剂/激活剂/正调节剂药物。为了减少跨人群分析中的潜在偏差,研究还使用仅包含欧洲人群的GWAS meta分析结果进行了复制分析。结果观察到高度的可重复性(Jaccard指数为0.667用于Banner药物MR,0.500用于ROSMAP药物MR,0.700用于BrainMeta药物MR),这表明PWAS结果的可靠性。研究进一步计算了跨转录组和蛋白组数据集的药物MR发现的一致性。结果显示,BrainMeta eQTL与ROSMAP pQTL MR分析之间的Jaccard指数为0.222,而ROSMAP和Banner pQTL MR分析之间的Jaccard指数为0.167。
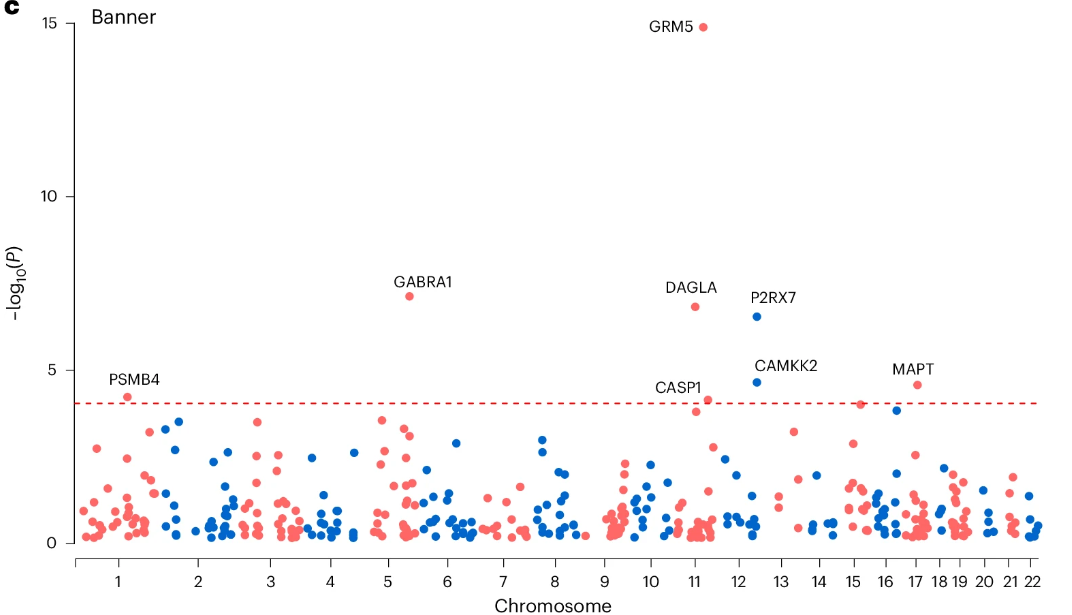
11、遗传学富集与基因集分析
研究表明,抑郁症的遗传性主要富集于大脑组织。特别地,抑郁症相关性在小脑半球和额叶皮层(BA9)中显示出最显著的富集,此外,在其他脑区也观察到显著的富集现象(假发现率(FDR)<0.05)。在细胞类型特异性的富集分析显示,兴奋性和抑制性神经元、神经母细胞以及少突胶质前体细胞在抑郁症的遗传学富集中具有显著性(Bonferroni校正后的P值阈值<1.28 × 10−3)。这些结果表明,神经元、神经母细胞和少突胶质前体细胞可能是抑郁症相关的重要细胞类型。
进一步的基因集富集分析通过MAGMA工具揭示了12个显著富集的基因本体(GO)条目,其中突触和突触后膜的富集最为显著。此外,神经分支形态发生的富集也表现出了显著性。这些分析结果强调了神经元和突触是抑郁症相关的关键细胞类型和生物学结构。
12、基因因果关系优先排序
为了识别最可能的因果基因,研究通过结合九种不同分析的证据,包括基因组位置、TWAS、PWAS、共定位、聚合遗传优先级评分(PoPS)、功能基因组学分析、eQTL分析以及基于总结数据的孟德尔随机化(SMR),进行了基因优先排序。研究采用了严格的Bonferroni校正,只有在多个校正后仍然显著的基因才被视为可靠的候选基因。因此,支持某基因的证据越多,越能表明该基因可能具有因果关系。
最终,研究共识别出40个位于32个抑郁症风险位点的基因,这些基因至少得到了五种分析方法的支持,其中8个基因得到了至少七种分析方法的支持。包括TMEM106B、AREL1、CTNND1、EPHB2、GIGYF2、PCDHA2、STAU1和TMEM258等基因,其中TMEM106B在所有分析中均被支持,并在这些基因中排名最高,表明该基因可能是抑郁症的因果基因。此外,CTNND1和AREL1也得到了八种证据的支持。值得注意的是,EPHB2还在药物孟德尔随机化分析中被优先排序为抑郁症的潜在治疗靶点。这些结果突出了抑郁症最可能的因果基因,并为进一步的功能表征和动物研究提供了重要的候选基因。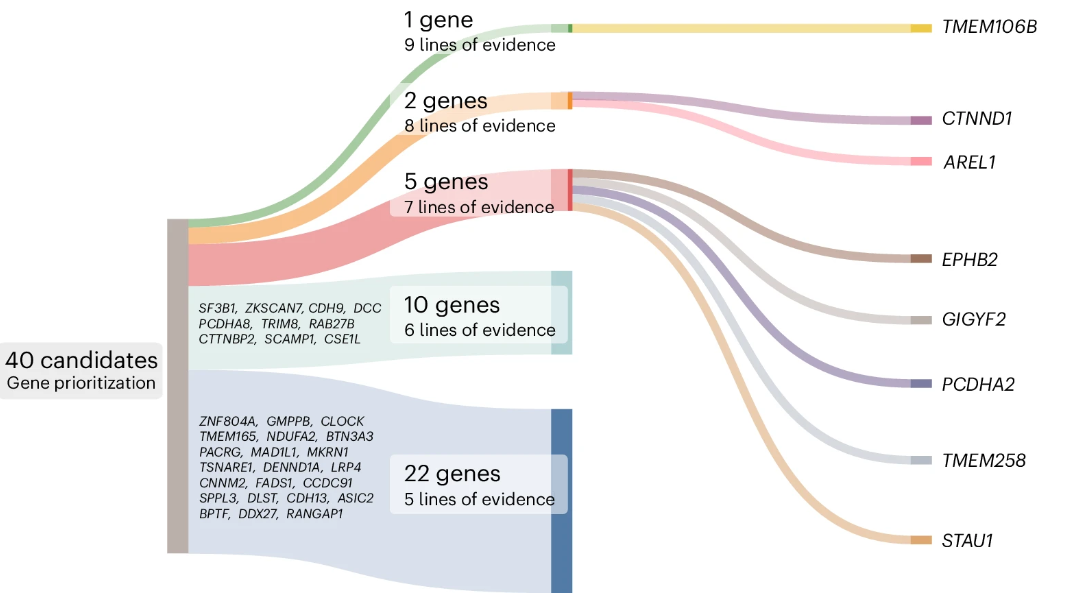
13、抑郁症风险基因在突触过程中的富集
基因优先排序分析识别出了40个高可信度的抑郁症风险基因,这些基因至少得到了五种分析方法的支持。研究使用了SynGO注释,评估了这些基因在突触中的作用。编码突触后特化(如CTNND1、ASIC2和DCC)和突触前本体术语(如CDH9、CTNND1、EPHB2、RAB27B和SCAMP1)的基因显示出显著的富集。
另外,九个基因与SynGO生物学过程注释相关,涉及突触信号传递、突触组织以及突触后和突触前过程。这些结果表明,抑郁症风险基因在突触过程中的富集具有重要意义。
14、Tmem106b基因敲低引起抑郁样行为
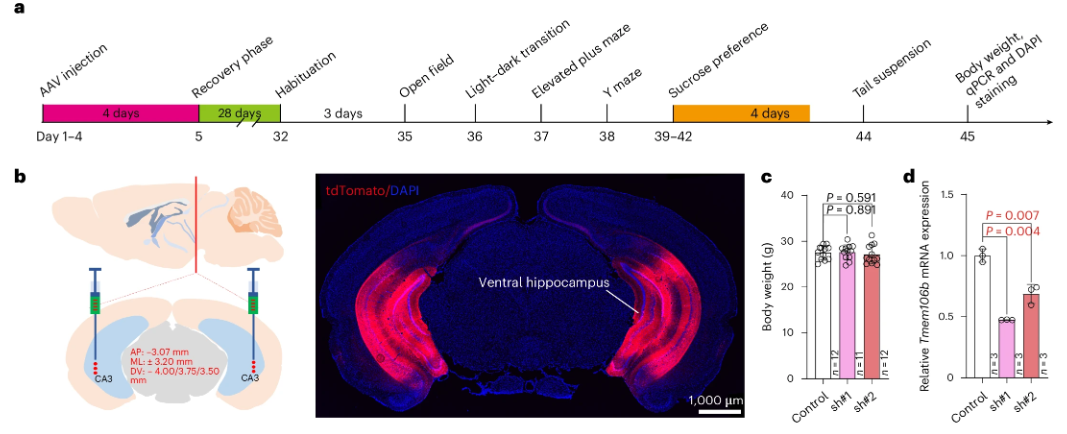
动物模型在验证由人类遗传研究识别的风险基因是否参与疾病发病机制中起着关键作用。前述分析表明,TMEM106B在抑郁症风险基因中排名靠前。为了进一步探究TMEM106B是否为真正的抑郁症风险基因,研究者在小鼠海马区敲低了Tmem106b(人类TMEM106B的同源基因,Ensembl ID:ENSMUSG00000029571),该大脑区域在小鼠模型和人类研究中已被报道与抑郁症密切相关,并且Tmem106b在此区域高度表达。研究者随后进行了系列行为学实验。
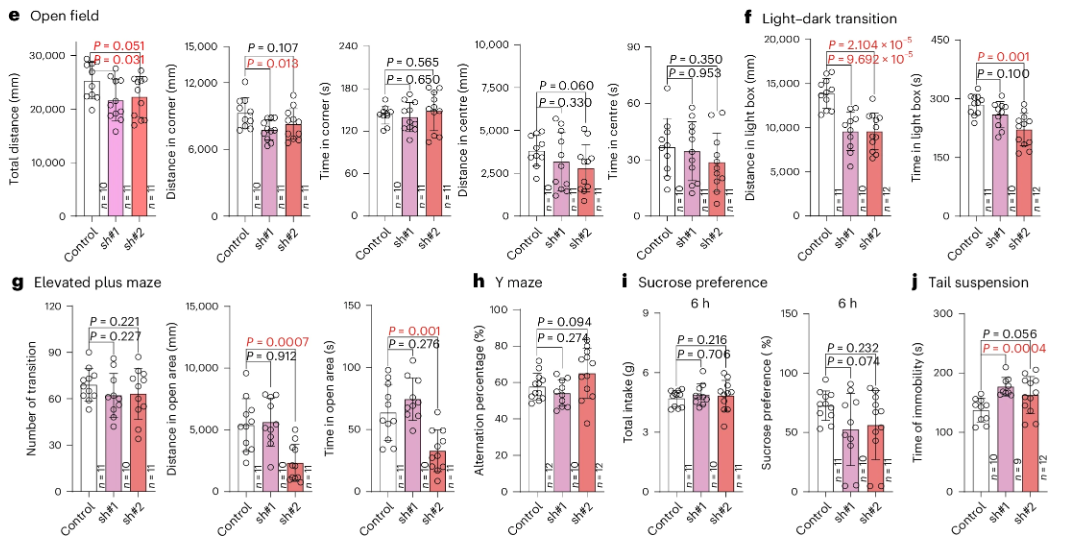
旷场测试结果表明,Tmem106b基因敲低小鼠表现出焦虑样行为。明暗转换实验显示,Tmem106b基因敲低小鼠更倾向于呆在暗箱中,并且在亮箱中的活动距离较对照组小。高架十字迷宫测试结果表明,Tmem106b基因敲低小鼠更偏好待在封闭臂,而非开放臂,表明这些小鼠表现出焦虑样行为。空间工作记忆测试(Y迷宫测试)表明,Tmem106b基因敲低小鼠的空间工作记忆与对照组无显著差异。进一步使用蔗糖偏好测试和尾悬挂测试评估抑郁样行为,结果发现Tmem106b基因敲低小鼠的蔗糖偏好未显著低于对照组,但其不动时间显著增加。
这些行为学结果表明,Tmem106b基因敲低导致了焦虑和抑郁样行为。综合来看,这些结果表明,在小鼠腹侧海马区敲低Tmem106b可以导致焦虑和抑郁样行为,这一发现强有力地支持了TMEM106B作为抑郁症风险基因的遗传学证据。

汇报人:黄石
导师:赵宇
审核:胥飞宇、庞文都、任建君
