


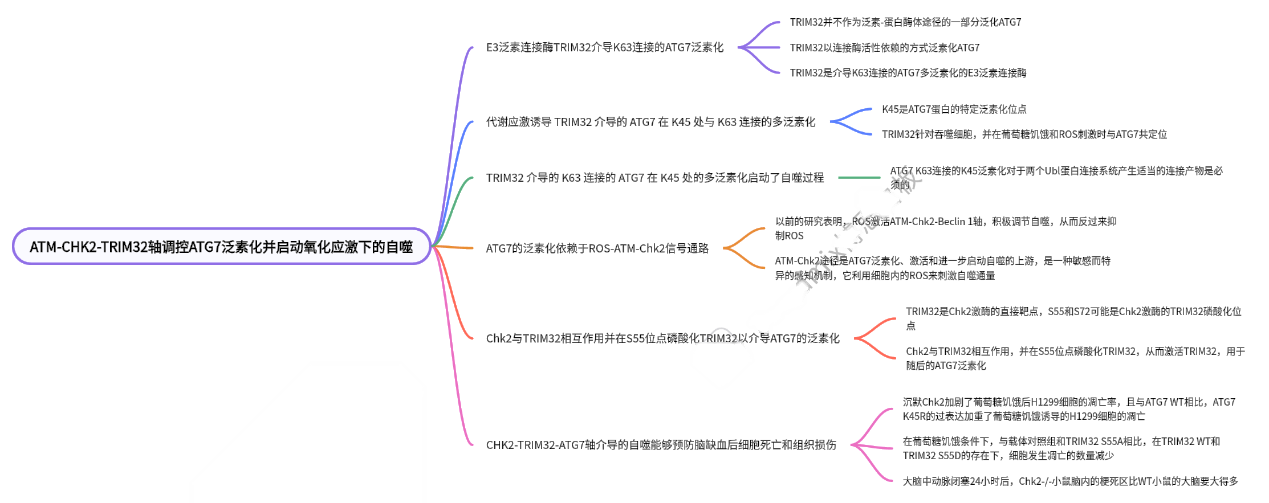
精读分享│【Cell Reports】:ATM-CHK2-TRIM32轴调控ATG7泛素化并启动氧化应激下的自噬
英文题目:ATM-CHK2-TRIM32 axis regulates ATG7 ubiquitination to initiate autophagy under oxidative stress
中文题目:氧化应激下ATM-CHK2-TRIM32轴调控ATG7泛素化启动自噬
期 刊:Cell Reports (IF=8.8)
单位:中国医科大学
时间:2023年11月28日

摘要:
氧化应激诱导的自噬有助于预防细胞损伤和维持内稳态。然而,启动自噬的调节途径仍未明晰。本团队之前展示了作为信号分子的活性氧(ROS)激活ATM-CHK2通路和促进自噬。本文中,团队发现E3泛素连接酶TRIM32作用在ATM-CHK2下游以调节ATG7泛素化。在代谢应激下,ROS在S1981位点诱导ATM磷酸化,从而在T68位点促进CHK2磷酸化。团队表明CHK2结合在S55位点并促使TRIM32磷酸化,然后在K45位点介导ATG7的K63连接泛素化以启动自噬。此外,卒中模型中Chk2-/-小鼠的梗塞表型表型加重、TRIM32磷酸化和ATG7泛素化减低。综上,团队提出了ROS通过ATM-CHK2-TRIM32-ATG7轴启动自噬,以维持细胞内稳态和保护暴露于病理条件下的细胞免受应激引起的组织损伤的分子机制。
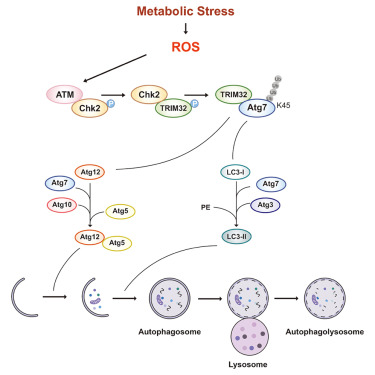
图形摘要
引言:
自噬是真核细胞中抵消营养耗竭和毒性蛋白应激条件的一种降解途径和保护性的分解代谢机制。在营养缺乏时,受损的细胞器通过自噬途径进行回收,产生对维持细胞稳态至关重要的大分子和能量。自噬还通过清除毒性蛋白质,预防其积累,保护了细胞。作为参与多种生理过程的活性氧(ROS)信号分子,也被发现具有促进自噬的作用。然而,ROS激活自噬的具体机制尚不清楚。
迄今为止,真核生物鉴定出了40多个自噬相关基因(ATG),其中约20个被认为核心ATG基因。细胞缺乏必要的自噬效应酶ATG7时表现出"自噬缺陷"。ATG7作为E1-类酶,在两个平行的泛素样( ubiquitin-like,Ubl)蛋白共轭系统中发挥作用,产生共轭复合物ATG8-PE和ATG12-ATG5。此外,ATG12-ATG5偶联物与Atg16一起作为E3连接酶,促进脂质磷脂酰乙醇胺(PE)与ATG8家族成员(LC3s/GABARAPs)的结合。这两个Ubl蛋白结合系统在自噬泡的扩展中起着至关重要的作用。当ATG7缺失时,ATG8的脂质化受阻,导致自噬泡膜的扩张受限,自噬体成熟受阻,自噬体不能与溶酶体结合,严重损害了自噬进程。尽管ATG7的自噬相关功能已被广泛研究,但ATG7激活发生的分子机制仍不清楚。
较多证据表明,ROS是自噬进程中的关键信号转导分子。例如,ROS诱导的ATG9A通过调节TRAF6和A20促进了Beclin1-VPS34-UVRAG复合体的组装,增加VPS34活性并促进其自噬。在先前研究中,作者揭示了CHK2以ROS依赖的方式磷酸化Beclin1,从而抑制Beclin1-Bcl2自噬复合体的形成并调节自噬。目前关于ROS与自噬之间的联系的研究主要集中在自噬的诱导和调控上,但尚未解答ROS是否对自噬的启动和进展产生直接作用的问题。
本研究鉴定了ATG7在K45位点的K63连接泛素化,这对于ATG7与Atg12或LC3的结合以及进一步的自噬体形成是必需的。值得注意地是,ROS是代谢应激下激活ATM-CHK2通路的关键信号分子,其中CHK2在S55位点磷酸化TRIM32 E3泛素连接酶以介导ATG7泛素化。本研究发现,代谢应激诱导的ROS累积通过磷酸化-泛素化级联反应激活ATM-CHK2-TRIM32-ATG7信号通路来启动自噬,阐述了一种ROS直接诱导自噬体形成和扩张的机制。ATM-CHK2-TRIM32-ATG7轴可能在自噬启动和保护细胞免受氧化应激中发挥重要作用。
总体研究思路:
主要结果:
1、E3泛素连接酶TRIM32介导ATG7的K63l连接泛素化
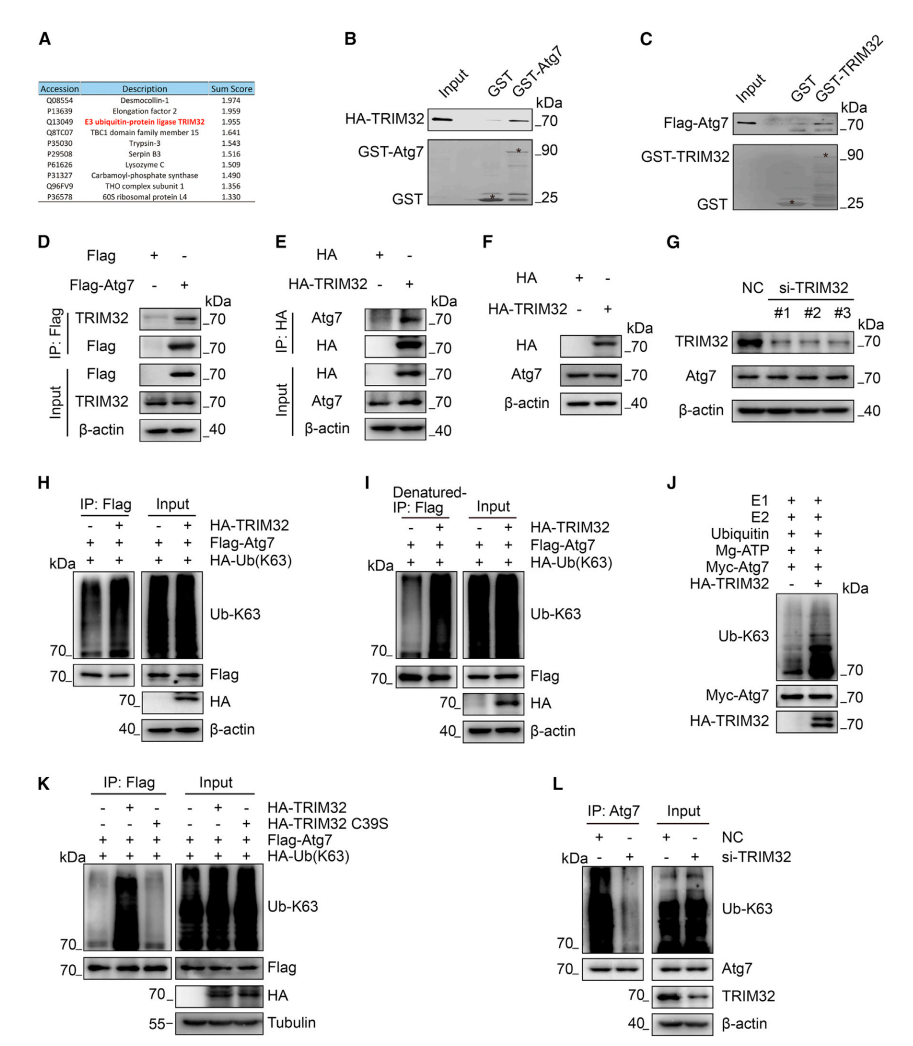
图1.E3泛素连接酶TRIM32介导K63连接的ATG7泛素化
(A)E3泛素蛋白连接酶TRIM32,用红色表示,通过质谱分析被鉴定为潜在的ATG7相互作用伴侣。
(B和C)GST拉下试验证明TRIM32与ATG7相互作用。将重组人TRIM32或ATG7与细菌表达的GST-ATG7或GST-TRIM32共同孵育。
(D)在转染后的HEK293细胞中进行FLAG-ATG7与内源性TRIM32的共免疫沉淀试验(CoIP)。FLAG空载作为对照组进行了转染。
(E)在转染后的HEK293细胞中进行HA-TRIM32与内源性ATG7的CoIP试验。HA空载作为对照组进行了转染。
(F和G)通过转染表达HA-TRIM32质粒或siRNA-TRIM32的3个目的序列后,对ATG7蛋白水平变化进行免疫印迹分析(Western blot,WB)。
(H) 将转染FLAG-ATG7、HA-K63泛素(一种除K63外的所有赖氨酸残基都突变为精氨酸的泛素结构,因此只能形成K63连接的Ub链)48小时后的HEK293细胞裂解,带或不带HA-TRIM32,接着用抗FLAG抗体进行免疫沉淀,然后用抗K63连接的泛素抗体进行WB试验。
(I)将转染FLAG-ATG7和HA-泛素K63,带或不带HA-TRIM32 48小时后的HEK293细胞裂解,通过变性免疫沉淀和WB试验检测ATG7泛素化。
(J)将Myc-ATG7和HA-TRIM32分别通过Myc磁珠和HA磁珠从转染Myc-ATG7和HA-TRIM32的HEK293细胞中取出,按设计方法进行体外泛素化实验。
(K) HEK293细胞经过转染FLAGb-ATG7、HA-泛素K63、HAb-TRIM32或HA-TRIM32 C39S 48h后裂解,用抗FLAG抗体进行免疫沉淀,再用抗K63连接的泛素抗体进行免疫印迹。
(L) HEK293细胞经转染NC(对照组)或sib-TRIM32 48h后裂解,用ATG7抗体进行免疫沉淀,再用抗K63-泛素抗体进行免疫印迹。
ATG7是自噬通路的关键启动因子,但导致其激活的信号转导级联尚不清楚。虽然翻译后修饰(PTMs)调节蛋白质功能的多个方面,但包括泛素化在内的PTMs对ATG7蛋白功能的贡献尚不清楚。为了鉴定ATG7潜在的E3泛素连接酶,研究者对纯化的ATG7蛋白组分进行了质谱分析,并鉴定出E3泛素连接酶TRIM32是潜在的ATG7相互作用蛋白(图1A)。GST下拉实验证实TRIM32与ATG7在体外直接结合(图1B和1C)。通过免疫共沉淀试验(CoIP)在HEK293和HCT116细胞中证实了TRIM32与ATG7的相互作用(图1D和1E)。
由于泛素化可以通过泛素-蛋白酶体途径靶向降解蛋白质,因此研究者检测了TRIM32过表达是否影响ATG7的稳态水平。过表达血凝素(HA)-TRIM32(图1F)和使用小干扰RNA(siRNA)瞬时沉默的TRIM32均未引起HCT116细胞中ATG7蛋白水平的显著变化(图1G)。综上,这些结果表明TRIM32并不参与部分泛素-蛋白酶体途径促进ATG7泛素化。
随后,研究者探究了TRIM32是否通过K63连接的泛素化途径以非蛋白酶体依赖的方式促进ATG7泛素化,TRIM32的过表达导致ATG7的K63连接泛素化增加(图1H)。变性条件下的泛素化实验也提示TRIM32介导了ATG7的泛素化(图1I)。值得注意的是,体外泛素化实验进一步证实了TRIM32在诱导ATG7泛素化中的作用(图1J)。有研究报道,在肌肉萎缩过程中,TRIM32能够以连接酶活性依赖的方式促进ULK1的自噬功能。接着,检测了TRIM32是否以同样的方式促进ATG7泛素化。结果显示,TRIM32野生型(WT)而非催化突变体C39S诱导ATG7泛素化,表明TRIM32以连接酶活性依赖的方式泛素化ATG7(图1K)。此外,在HEK293和HCT116细胞通过siRNA瞬时沉默TRIM32降低了ATG7的K63连接的泛素化(图1L)。因此,这些结果表明TRIM32作为E3泛素连接酶介导ATG7的K63连接的多聚泛素化。
2、代谢应激诱导TRIM32介导的ATG7K63在K45位点的K63连接多聚泛素化的
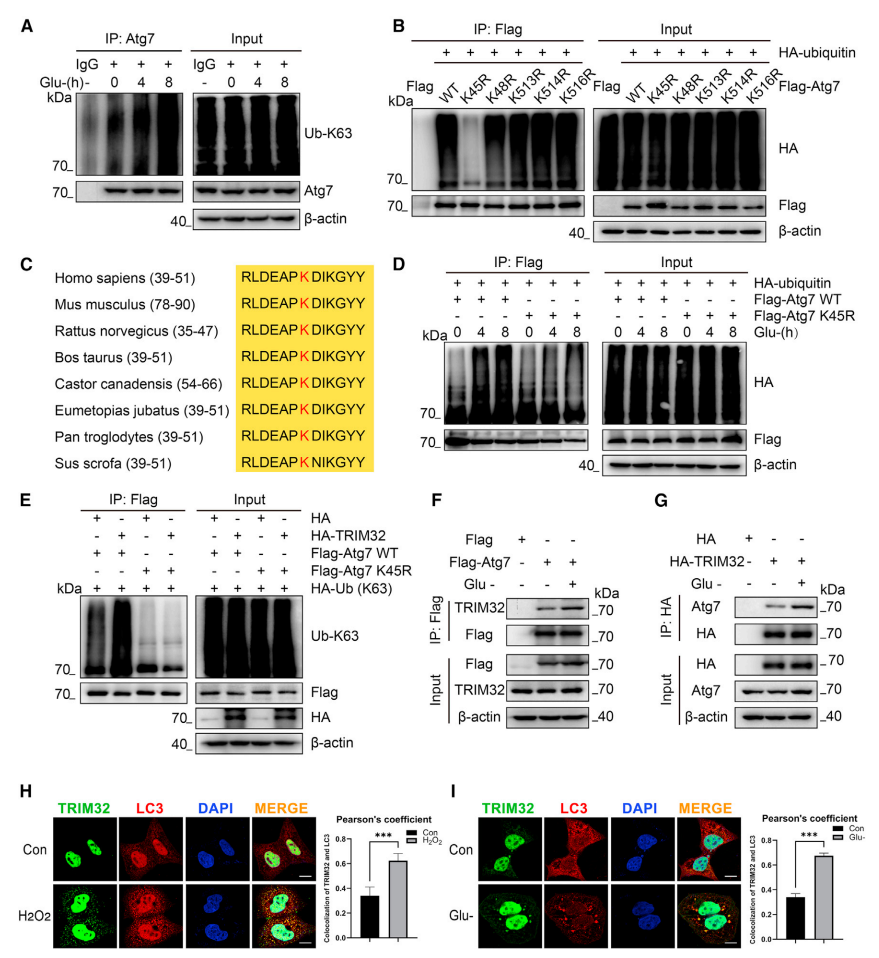
图2. 代谢应激诱导TRIM32介导的ATG7在K45位点的K63连接多泛素化。
(A)从用或不用无糖培养基处理的HCT116细胞中制备的裂解物,在指定时间后使用抗ATG7抗体或IgG对照物进行免疫沉淀,然后用抗K63连接泛素抗体进行免疫印迹分析。
(B)将转染HA泛素和指定的FLAG-ATG7构建体48小时后的HEK293细胞裂解,然后使用抗FLAG抗体进行免疫沉淀,随后用抗HA抗体进行免疫印迹分析。
(C)不同物种中ATG7同源物K45残基周围的序列比对。红色字母“K”表示ATG7的45位赖氨酸残基。
(D)将HA-泛素和FLAG-ATG7WT或FLAG-ATG7K45R分别转染至HEK293细胞。在无糖培养液中处理一定时间后,细胞裂解,用抗FLAG抗体进行免疫沉淀,然后用抗HA抗体进行免疫印迹。
(E) HEK293细胞经过仅用HA-泛素K63与HA或HA-TRIM32和FLAG-ATG7 WT或FLAG-ATG7 K45R共同转染后48h裂解,用抗FLAG抗体进行免疫沉淀,然后用抗K63连接的泛素、FLAG和HA抗体进行免疫印迹。
(F和G) 在含糖培养基和葡萄糖耗竭后培养,对内源性TRIM32与外源性FLAG-ATG7之间,以及内源性ATG7与外源性HA-TRIM32之间的相互作用进行CoIP分析。
(H) HEK293细胞用双氧水(500 μM)处理1h后,使用免疫荧光和定量分析进行TRIM32和LC3的共定位分析(比例尺,10μM)。计算皮尔逊系数以定量分析TRIM32和LC3的共定位。数据以六次独立实验的平均值±标准误差(SEM)表示;*p<0.001(Student t检验)。
(I) HEK293细胞经过葡萄糖耗竭培养1h后,TRIM32和LC3共定位进行免疫荧光和定量分析。计算皮尔逊系数以量化TRIM32和LC3的共定位(比例尺,10 μM)。数据以六次独立实验的平均值±SEM表示;*p<0.001(Student t检验)。
为了证实ATG7的K63连接的泛素化修饰在促进自噬的条件下发生,作者寻找了ATG7的K63连接的泛素化水平在代谢应激条件下的变化。首先,研究者发现在葡萄糖剥夺刺激下,ATG7的K63连接的泛素化水平逐渐增加(图2A)。同样,H2O2也诱导ATG7的K63连接的泛素化水平随着刺激时间的延长而逐渐升高。
研究者使用筛选每个潜在的赖氨酸以确定特定蛋白质的泛素化位点的泛素位点质谱分析来鉴定被TRIM32泛素化的ATG7上的赖氨酸残基。结果表明,K45、K48、K513、K514和K516均为ATG7蛋白潜在的泛素化位点。CoIP试验表明,当赖氨酸(K)K45被精氨酸(R)取代时,与在K48,K513,K514或K516位点的替换相比,泛素化水平显著降低(图2B),这表明K45是ATG7蛋白的特异性泛素化位点。值得注意的是,ATG7的K45位点在不同物种间高度保守(图2C)。此外,当将赖氨酸(K)K45替换为精氨酸(R)时,无论是葡萄糖耗竭培养还是TRIM32过表达,均不会导致HEK293和HCT116细胞的ATG7的K63连接的泛素化增加(图2D、2E)。这强烈表明K45位点是一个与生理相关的泛素化位点。
在启动自噬的条件下,TRIM32介导的ATG7泛素化预计会增加TRIM32和ATG7相互作用。为了检验这一点,研究者提出疑问:在代谢应激条件下,TRIM32和ATG7之间的相互作用是否受到影响。事实上,在葡萄糖耗竭培养下,研究者观察到TRIM32和ATG7之间的相互作用增强(图2F和2G)。此外,在H2O2刺激(图2H)和葡萄糖剥夺(图2I)的条件下,TRIM32与LC3和ATG7共定位,表明TRIM32靶向自噬细胞并与ATG7共定位以适应葡萄糖耗竭和ROS刺激条件。
3、TRIM32介导的K63连接的ATG7在K45位点的多聚泛素化启动自噬过程 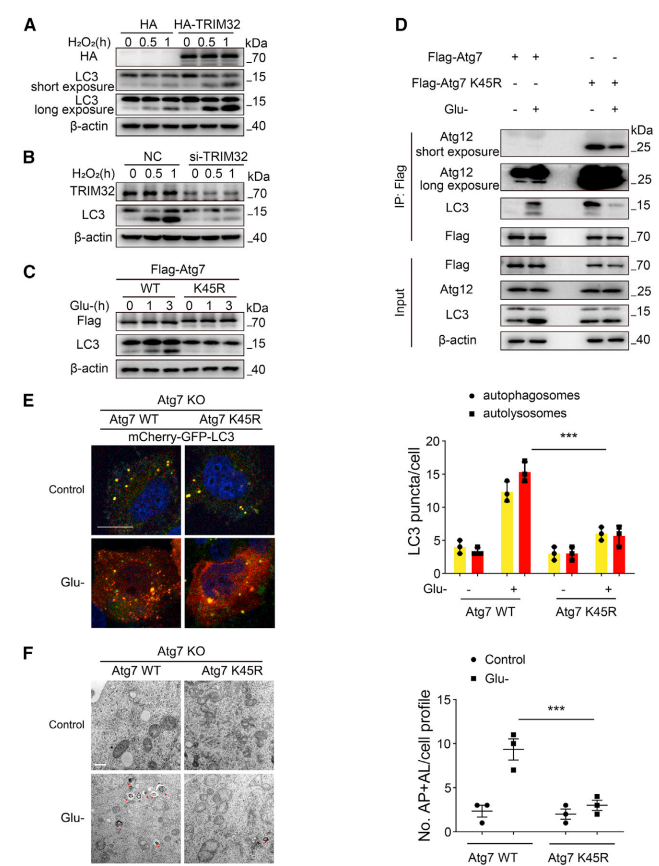
图3.TRIM32 介导的 K63 链接的 ATG7 在 K45 处的多泛素化启动了自噬过程
(A)HEK293细胞分别转染HATRIM32或HA 48h作为对照组,经H2O2(500 mM)刺激一定时间后裂解细胞,用特异性抗体免疫印迹法检测细胞中LC3和HA的表达水平。β-actin作为载样对照。
(B) HEK293细胞分别转染阴性对照或TRIM32特异性siRNA 48小时后,在H2O2(500 mM)刺激一定时间后裂解细胞,用特异性抗体免疫印迹法检测细胞中LC3和TRIM32的表达水平。β-actin作为载样对照
(C) HEK293细胞分别转染FLAG-ATG7WT或FLAG-ATG7K45R 48h后,在缺糖条件刺激一定时间后裂解,用特异性抗体免疫印迹法检测细胞中LC3和ATG7的表达。
(D)CoIP分析内源ATG12/LC3和外源FLAG-ATG7 WT/FLAG-ATG7 K45R在含糖培养基和葡萄糖耗竭后培养的相互作用。
(E)代表性共聚焦显微镜图像显示:在葡萄糖耗竭培养1h后,敲除ATG7基因的HEK293细胞经过ATG7 WT或ATG7 K45R转染后稳定表达GFP-mCherry-LC3的自噬通量。(比例尺,10μM)。在一定时间内,对葡萄糖耗竭培养后的自噬小体(黄色)和自溶酶体(红色)LC3斑点进行定量分析。数据以三次独立实验的平均值±SEM表示;*p<0.001(Student t检验)。
(F)代表性透射电镜图像显示:在正常培养基或无糖培养基中培养1h的ATG7基因敲除的HEK293细胞中转染ATG7 WT或ATG7 K45后的典型自噬小泡或自噬体。红色箭头表示双膜自噬结构(比例尺,1μM)。定量电镜分析在正常培养基或无糖培养基中培养的转染ATG7 WT或ATG7 K45R基因的HEK293细胞。数据以三次独立实验的平均值±SEM表示;*p<0.001(Student t检验)。
虽然葡萄糖耗竭培养或H2O2诱导的自噬与ATG7的K63连接的泛素化增加有关,而ATG7是维持自噬正常进展所必需的蛋白,但尚不清楚ATG7在K45位点的K63连接泛素化是否为启动自噬所必需的过程。研究人员发现TRIM32过表达促进自噬,表现为LC3的非脂质化形式(LC3-I)转化为磷脂酰乙醇胺结合形式(LC3-Ⅱ) (图3A)。相反,siRNAs瞬时沉默TRIM32抑制自噬后,LC3-I向LC3-Ⅱ的转化减少(图3B)。此外,研究人员发现当ATG7的K45位点被精氨酸取代且该位点无法被泛素化时,由葡萄糖耗竭、H2O2或EBSS诱导的自噬显著抑制(图3C)。重要的是,在代谢应激中,与ATG7 WT相比,ATG7 K45R突变体与LC3和ATG12的相互作用被破坏(图3D),这表明ATG7在K45位点的K63连接泛素化对于两种Ubl蛋白接合系统产生适当的接合产物是必不可少的。
当赖氨酸(K)K45替换为精氨酸(R)时,应对代谢应激的自噬流也被显著抑制(图3E)。细胞内LC3蛋白点状结构提示,在葡萄糖耗竭培养下,稳定表达mCherry-GFP-LC3的ATG7敲除的HEK293细胞经过转染ATG7 K45R而不是ATG7 WT时,自噬体的形成明显减少。最终,定量电镜分析显示,相比于表达ATG7 WT的细胞,表达ATG7 K45R的ATG7敲除细胞表现出代谢应激诱导的自噬结构(自噬小体和自噬溶酶体) 减少 (图3F)。WB检测ATG7敲除的HEK293细胞中ATG7 WT和ATG7 K45R的蛋白水平。综上,这些发现表明TRIM32对ATG7在K45位点的K63连接泛素化在代谢应激下启动自噬过程中起着重要的作用。
4、ATG7的泛素化依赖于ROS-ATM-CHK2信号通路
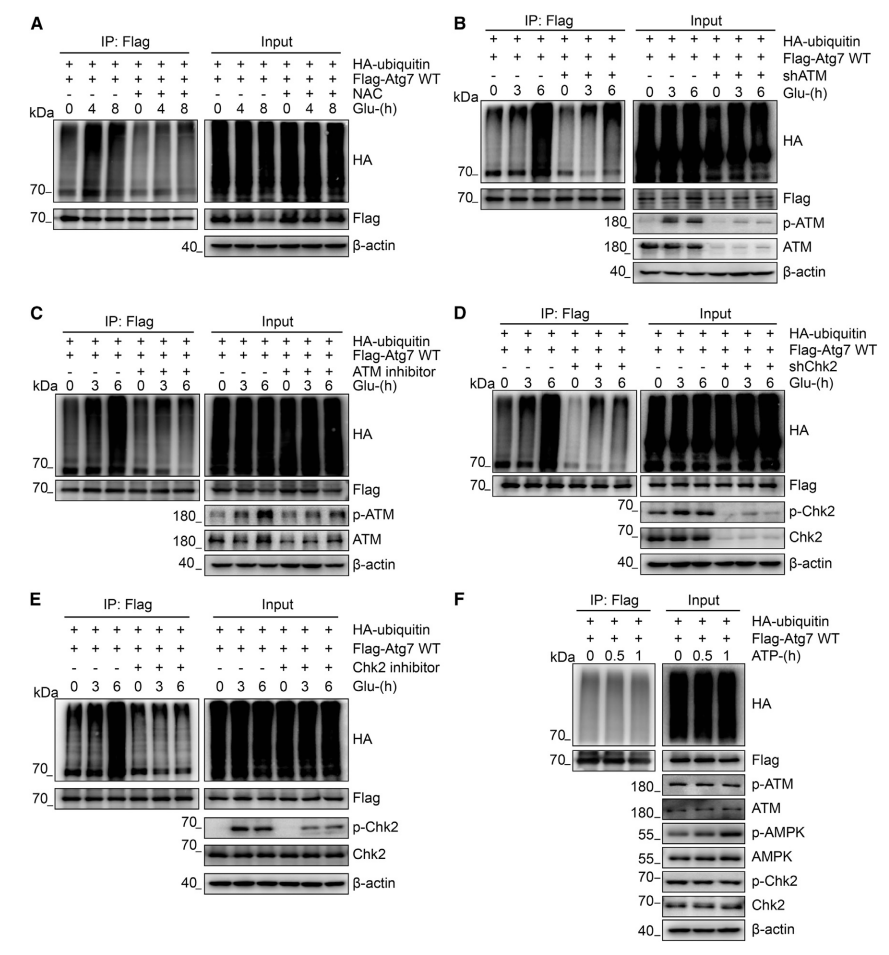
图4.ATG7的泛素化依赖于ROS-ATM-Chk2信号通路
(A)将HA-泛素和FLAG-ATG7WT转染入HCT116细胞,用或不加NAC(3 mM,4h)处理细胞。细胞在含或不含无糖培养基的条件下处理一段时间后裂解,然后用抗FLAG抗体进行免疫沉淀,用抗HA抗体进行免疫印迹分析泛素水平。
(B和D)将稳定表达或不表达ATM或Chk2 shRNA的H1299细胞分别与HA-泛素和FLAG-ATG7 WT共转染48h后,在含或不含无糖培养基的条件下处理细胞一段时间,然后裂解细胞,用抗FLAG抗体进行免疫沉淀,然后用抗HA抗体进行免疫印迹分析泛素水平。
(C和E)将转染HA-泛素和FLAG-ATG7 WT的HCT116细胞分别用加或不加ATM(1:1000)抑制剂或Chk2(1:1000)抑制剂处理。细胞在含或不含无糖培养基的条件下处理一段时间后裂解,然后用抗FLAG抗体进行免疫沉淀,用抗HA抗体进行免疫印迹分析泛素水平。
(F)将HA-泛素和FLAG-ATG7WT转染入HEK293细胞。在2-脱氧-D-葡萄糖(2DG)(5mM)和寡霉素(2.5μM)处理一定时间后裂解细胞,然后用抗FLAG抗体进行免疫沉淀,用抗HA抗体进行免疫印迹分析泛素水平。利用输入样本进行WB分析,发现p-ATM Ser1981、ATM、p-Chk2 Thr68、Chk2、p-AMPKa Thr172和AMPKa。
营养剥夺和代谢应激会引起ROS产物的积累。前期研究表明,ROS激活ATM-CHK2-Beclin 1轴正向调控自噬,进而抑制ROS。因此,研究人员想知道ATG7的泛素化是否依赖于ROS-ATM-CHK2途径。为了检测这一点,研究人员用自由基清除剂N -乙酰半胱氨酸(NAC)处理细胞,发现葡萄糖耗竭诱导的ATG7泛素化减少(图4A)。这一结果表明ROS产物是关键的信号分子。
为了进一步确认共济失调毛细血管扩张突变(ATM)在ATG7泛素化中的作用,研究人员检测了短发夹状RNA(shRNA)诱导的ATM稳定敲低和抑制ATM活性对ATG7泛素化的影响。ATM的稳定敲低和药理性ATM抑制剂的应用均减弱了葡萄糖耗竭时ATG7泛素化增加的表型(图4B和4C)。此外,在shCHK2处理和药理性Chk2抑制后的细胞中,ATG7泛素化的增加也受到抑制(图4D和4E)。此外,用2DG和寡霉素的干预诱导能量耗竭激活AMPKα1/2而不影响ATM/CHK2通路,表现为ATG7的泛素化水平无明显变化(图4F)。这些结果表明,是一种利用细胞内ROS刺激自噬通量的敏感而特异的感知机制——ATM-CHK2通路,通过ATG7泛素化、激活和进一步启动自噬的上游通路。
5、CHK2与TRIM32相互作用,在S55位点磷酸化TRIM32以介导ATG7泛素化
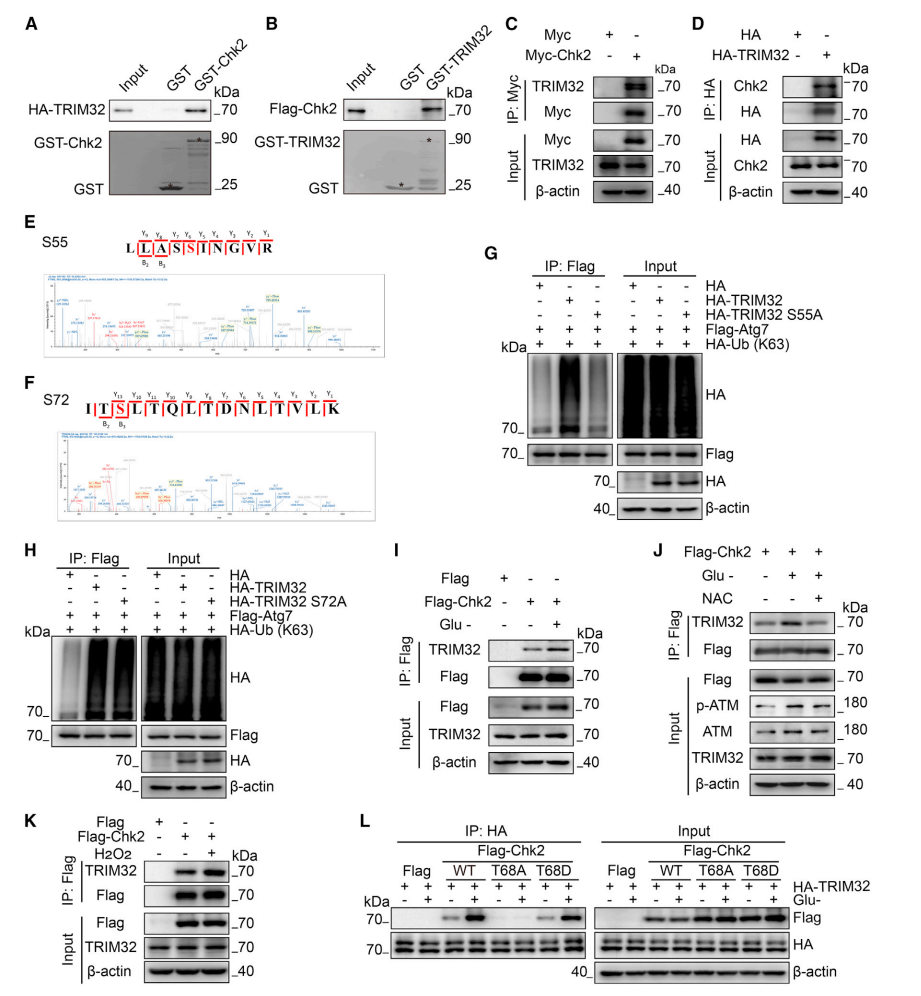
图5.Chk2与TRIM32相互作用,在S55位点磷酸化TRIM32以介导ATG7泛素化
(A和B)通过GST下拉试验分析TRIM32与Chk2的相互作用。将重组人TRIM32或CHK2与细菌表达的GST-CHK2或GST-TRIM32共孵育。
(C)CoIP分析转染Myc-Chk2和内源性TRIM32的HEK293细胞。以MYC空载为对照。
(D)CoIP分析转染HA-TRIM32和内源性Chk2的HEK293细胞。以HA空载为对照。
(E和F)质谱图显示了TRIM32蛋白S55和S72的磷酸化位点。
(G和H)将仅HA-K63泛素化、FLAG-ATG7和HA-TRIM32野生型或突变体(HA-TRIM32 S55A或HA-TRIM32 S72A)共转染至HEK293细胞。以HA空载为对照。48h后裂解细胞,用抗FLAG抗体进行免疫沉淀,然后用抗K63连接的泛素、FLAG和HA抗体进行免疫印迹分析。
(I)CoIP分析内源TRIM32和外源FLAG-Chk2在含糖培养基和葡萄糖耗竭后培养的相互作用。
(J)CoIP分析内源TRIM32和外源FLAG-Chk2在含糖培养基和葡萄糖耗竭后培养经NAC(3mM)处理后的相互作用。
(K) CoIP分析用或不用H2O2(500μM)处理1h后的内源TRIM32与外源FLAG-Chk2的相互作用。
(L) CoIP分析将HA-TRIM32和FLAG空载、FLAG-CHK2WT、T68A和T68D分别转染入HEK293细胞。细胞经含或不含无糖培养基培养后,裂解细胞,用抗HA抗体进行免疫沉淀,然后用抗FLAG和HA抗体进行免疫印迹。
由于发现了ATG7的泛素化是由ROS-ATM-CHK2途径介导的,而CHK2是一种被磷酸化的激酶,因此研究者探究CHK2是否通过磷酸化E3泛素连接酶TRIM32来促进ATG7的泛素化。为了验证这一点,研究者首先通过GST下拉实验检测了CHK2与TRIM32在体外的相互作用(图5A和5B)。在HEK293和HCT116细胞中通过CoIP实验进一步证实CHK2与TRIM32之间存在显著的相互作用(图5C、5D)。这一结果有力证明TRIM32是CHK2激酶的直接靶标。
为了确定CHK2磷酸化TRIM32的位点,研究人员进行了CHK2激酶实验的磷酸化位点质谱分析。S55和S72被鉴定为CHK2激酶可能的TRIM32磷酸化位点(图5E和5F)。接下来,研究人员探究了CHK2诱导的TRIM32磷酸化是否决定了其E3泛素连接酶活性。将TRIM32的S55替换为丙氨酸(A)后,在HEK293(图5G)和HCT116细胞中,ATG7的泛素化水平与TRIM32 WT相比均显著降低。然而,在HEK293(图5H)和HCT116细胞中,与TRIM32 WT相比,过表达TRIM32 S72A仍然促进了ATG7的泛素化。因此,S55似乎是CHK2激酶激活TRIM32的相关磷酸化位点。
最后,研究人员检测了CHK2和TRIM32在代谢应激下的相互作用。正如预期的那样,当葡萄糖耗竭时,CHK2和TRIM32之间的相互作用增强,并且被自由基清除剂NAC抑制(图5I-5J)。这些结果以及CHK2和TRIM32在H2O2刺激下相互作用的增加(图5K),表明CHK2和TRIM32的相互作用在葡萄糖耗竭时以依赖ROS的方式增强。值得注意的是,无论葡萄糖是否耗竭,CHK2 T68A几乎不与TRIM32相互作用。然而,在葡萄糖耗竭后,活化的CHK2 T68D形式与TRIM32的相互作用增强,与WT CHK2间作用类似(图5L)。综上所述,这些发现表明CHK2与TRIM32相互作用,并在S55位点磷酸化TRIM32,从而激活TRIM32进行随后的ATG7泛素化。
6、CHK2-TRIM32-ATG7轴介导的自噬能够预防脑缺血后的细胞死亡和组织损伤
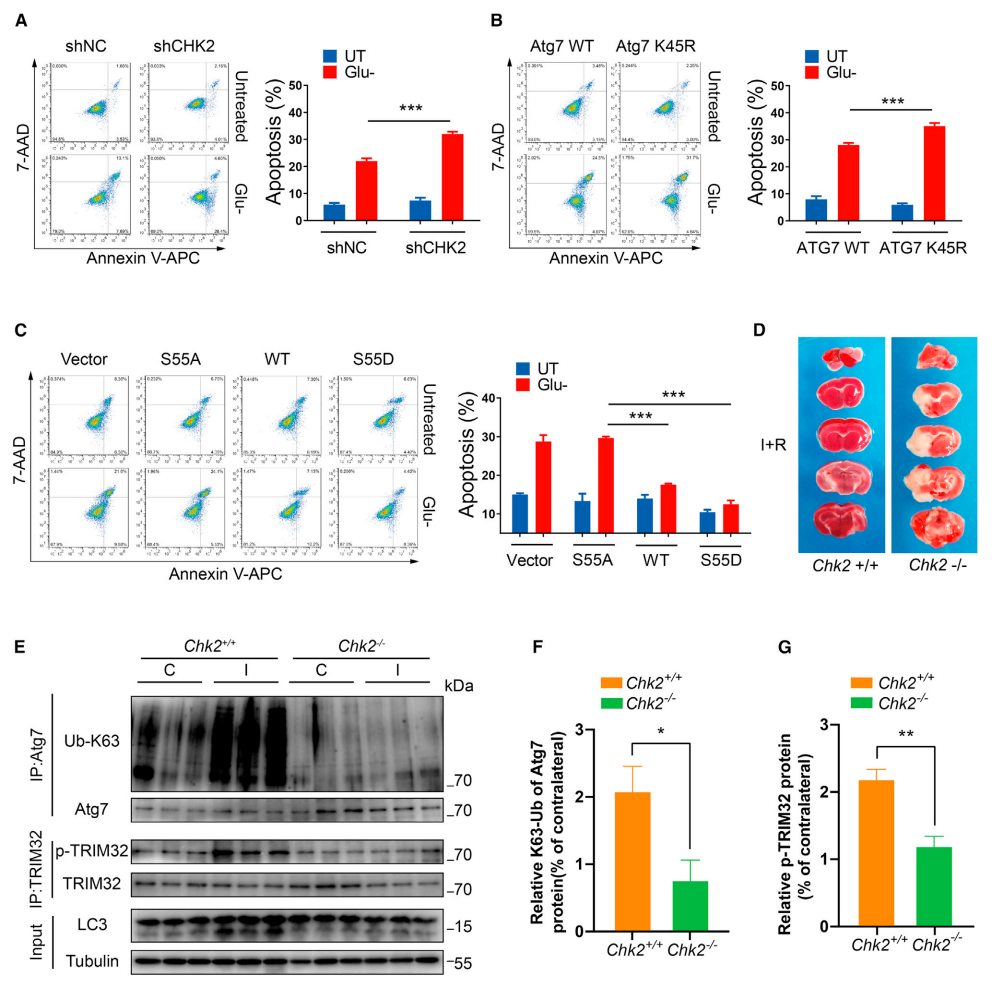
图6.Chk2-TRIM32-ATG7轴介导的自噬可防止脑缺血后的细胞死亡和组织损伤
(A) 在含糖培养基或葡萄糖耗竭16小时后培养下,将空载或Chk2 shRNA转染至H1299细胞。代表性的荧光激活细胞分选(FACS)分析凋亡情况。数据以三次独立实验的平均值±SEM表示;*p<0.001(Student t检验)。
(B) 在含糖培养基或葡萄糖耗竭16小时后培养下,将FLAG-ATG7WT或FLAG-ATG7 K45R表达载体转染入H1299细胞。代表性的流式细胞分析凋亡情况。数据以三次独立实验的平均值±SEM表示;*p<0.001(Student t检验)。
(C) 在含糖培养基或葡萄糖耗竭16小时后培养下,将HA、HA-TRIM32 WT、HA-TRIM32 S55A或HA-TRIM32 S55D表达载体分别转染入H1299细胞。代表性的流式细胞分析凋亡情况。数据以三次独立实验的平均值±SEM表示;*p<0.001(Student t检验)。
(D)Chk2+/+和Chk2-/-小鼠大脑中动脉结扎1h,再灌注24h,对侧(C)和同侧(I)脑组织进行2% 2,3,5-三苯基四氮唑(2% 2,3,5-TTZ)染色。
(E-G)免疫沉淀法和免疫印迹法检测脑缺血和再灌流后Chk2+/+和Chk2-/-鼠皮质提取物中ATG7、Ub-K63、TRIM32和p-TRIM32以及LC3水平。量化p-TRIM32和K63连接的ATG7蛋白泛素化水平(小鼠n=3)。数据以均值±SEM表示;*p<0.05,**p<0.01(Student t检验)。
为了进一步探究CHK2-TRIM32-ATG7的生理功能,研究人员进行了葡萄糖耗竭情况下的细胞凋亡实验。结果表明,CHK2沉默加重了H1299细胞在葡萄糖耗竭后的凋亡率(图6A)。此外,与ATG7 WT相比,过表达ATG7 K45R加重了葡萄糖耗竭诱导的H1299细胞凋亡(图6B)。此外,为了检测CHK2介导的TRIM32磷酸化是否参与了这一现象,研究了TRIM32非磷酸化突变体S55A和磷酸化模拟突变体S55D在葡萄糖耗竭培养下对细胞凋亡的影响。结果表明,在葡萄糖耗竭培养下,与空载体对照和TRIM32 S55A相比,TRIM32 WT和TRIM32 S55D存在时,凋亡细胞数量减少(图6C)。
由于脑缺血可以引起ROS诱导的组织损伤,并伴随一系列的代谢事件。因此研究人员在小鼠体内卒中模型中研究了CHK2-TRIM32-ATG7轴的作用。在Chk2基因敲除(Chk2-/-)小鼠中进行了大脑中动脉闭塞(MCAO)实验。在MCAO 24小时后,Chk2-/-小鼠的大脑梗死面积远远大于WT小鼠(图6D)。此外,与对侧大脑相比,WT小鼠患侧大脑区域的TRIM32磷酸化和ATG7泛素化水平增加。然而,Chk2-/-小鼠的无明显变化(图6E-6G)。以上数据表明CHK2-TRIM32-ATG7轴可能有助于缺血性脑卒中时的脑细胞存活。
总结:
氧化应激诱导的自噬有助于防止细胞损伤和维持内环境稳定。然而,启动自噬的调控途径尚不清楚。作者团队之前的研究表明,活性氧作为信号分子激活ATM-CHK2通路,促进自噬。本研究发现E3泛素连接酶TRIM32作用于ATM - CHK2下游,调控ATG7泛素化。在代谢应激条件下,ROS诱导ATM在S1981位点磷酸化,进而使CHK2在T68位点磷酸化。CHK2在S55位点结合并磷酸化TRIM32,然后介导ATG7在K45位点的K63连接泛素化从而启动自噬。此外,在卒中模型中Chk2-/-小鼠的梗塞表型加重,TRIM32的磷酸化和ATG7的泛素化水平降低。本研究提出了ROS通过ATM-CHK2-TRIM32-ATG7轴启动自噬的分子机制,以维持细胞内稳态,并保护暴露于病理条件下的细胞免受应激诱导的组织损伤。

汇报人:郝智贞
导师:刘锋、刘吉峰
审核:王欣怡、孙晓茹、任建君
