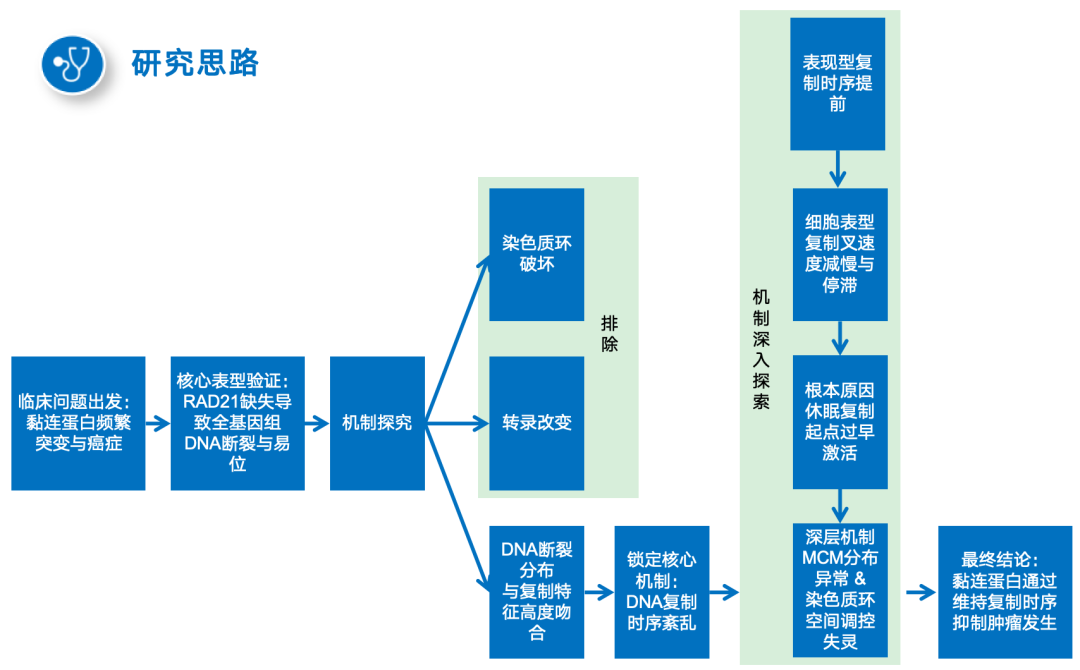
精读分享│【Nature Genetics】:黏连蛋白通过维持复制时序来抑制癌基因上的DNA损伤
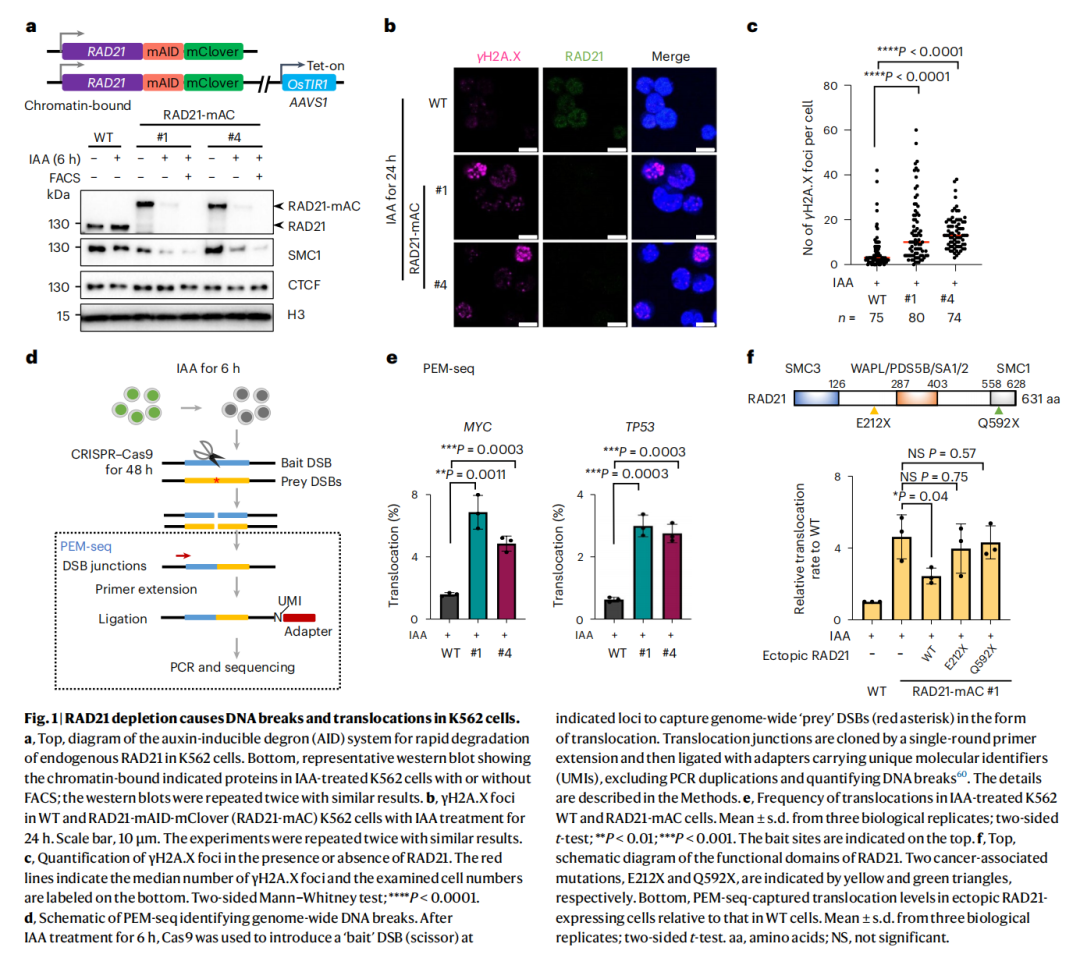
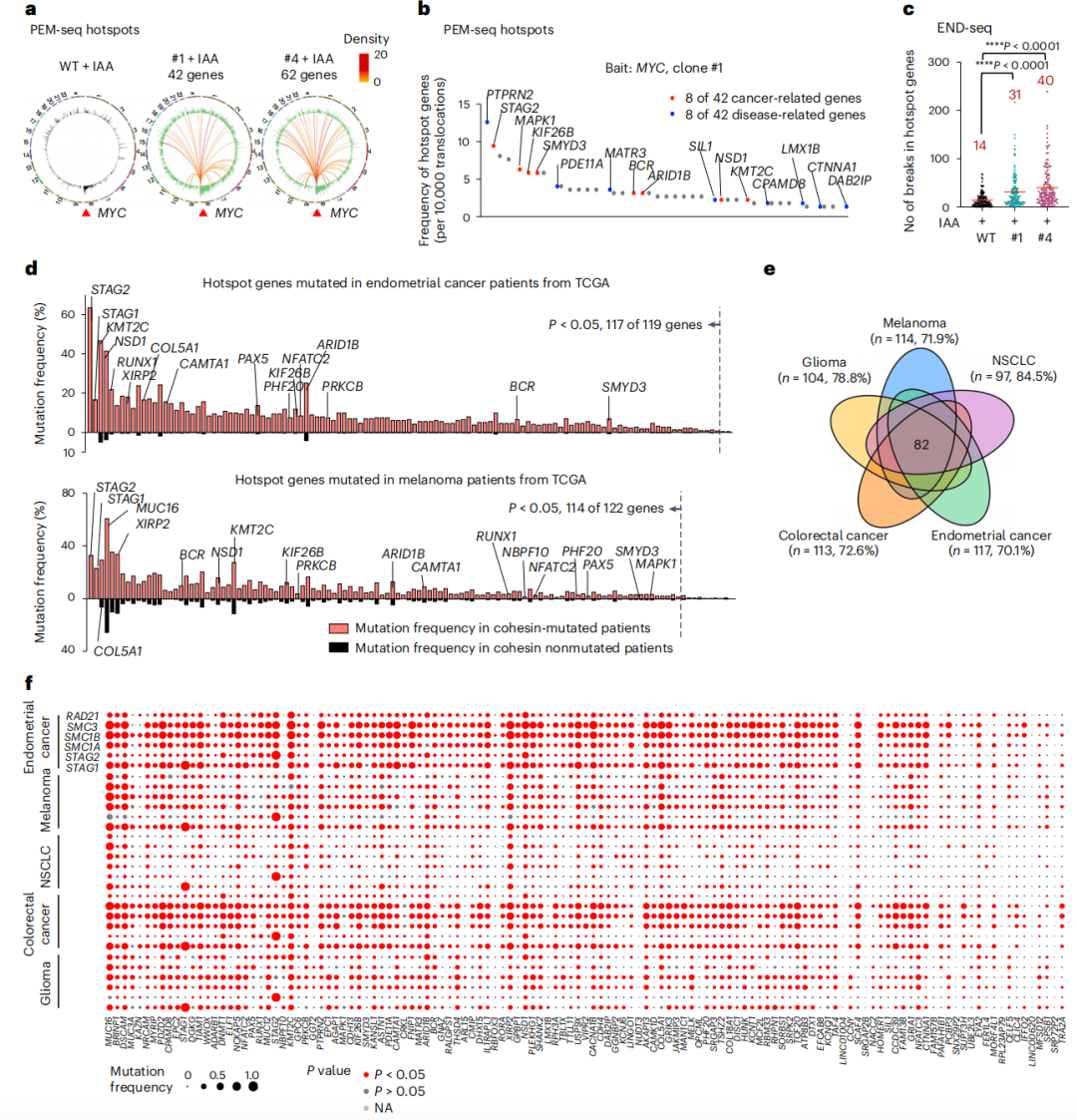
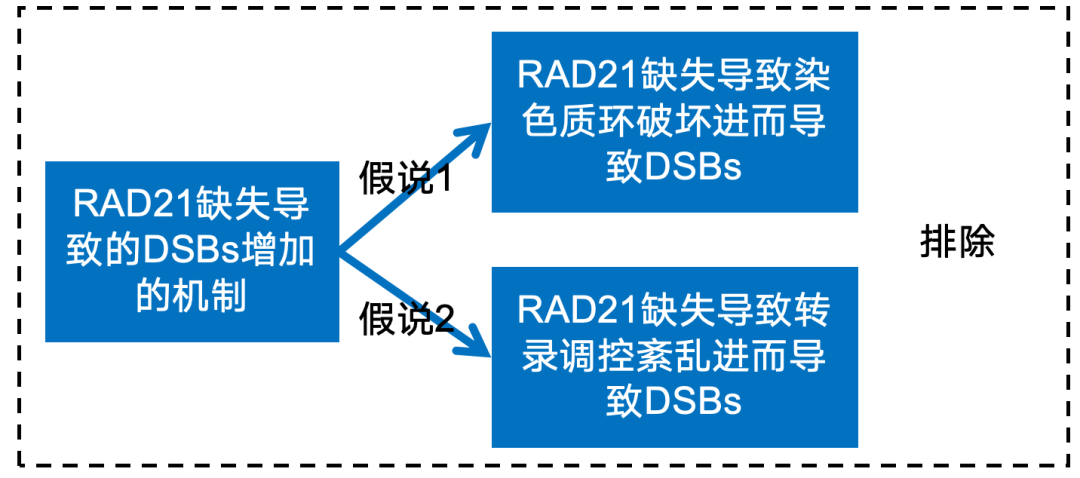
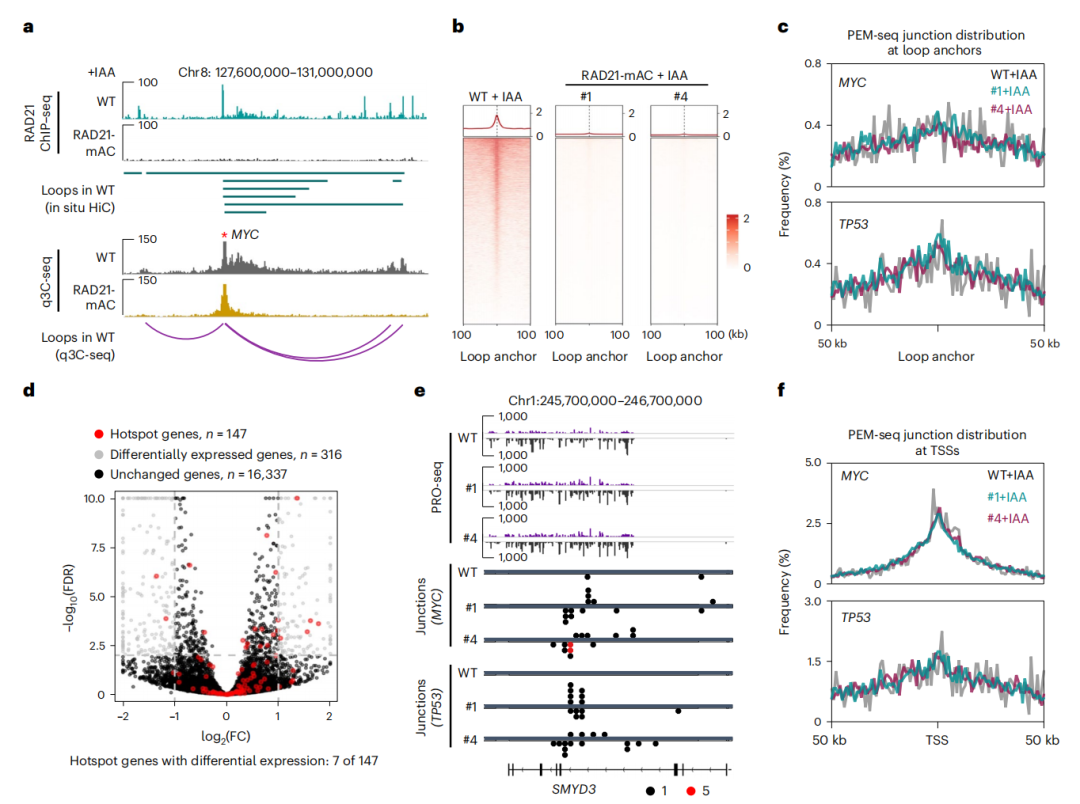
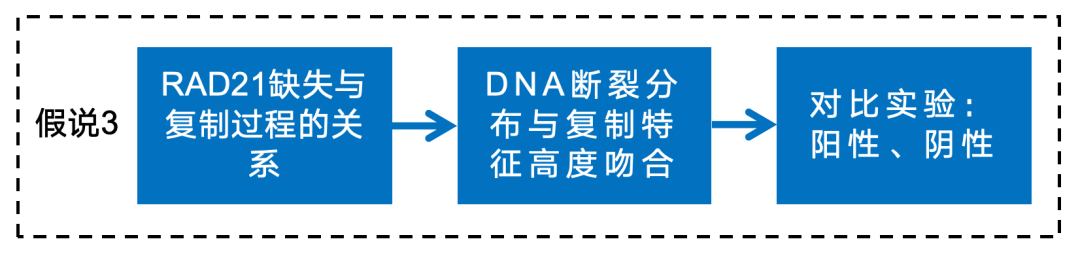
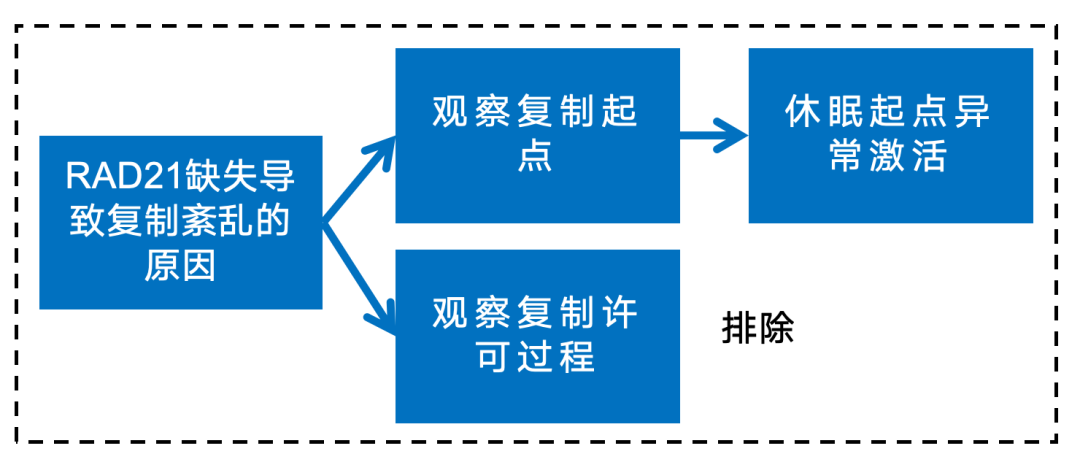
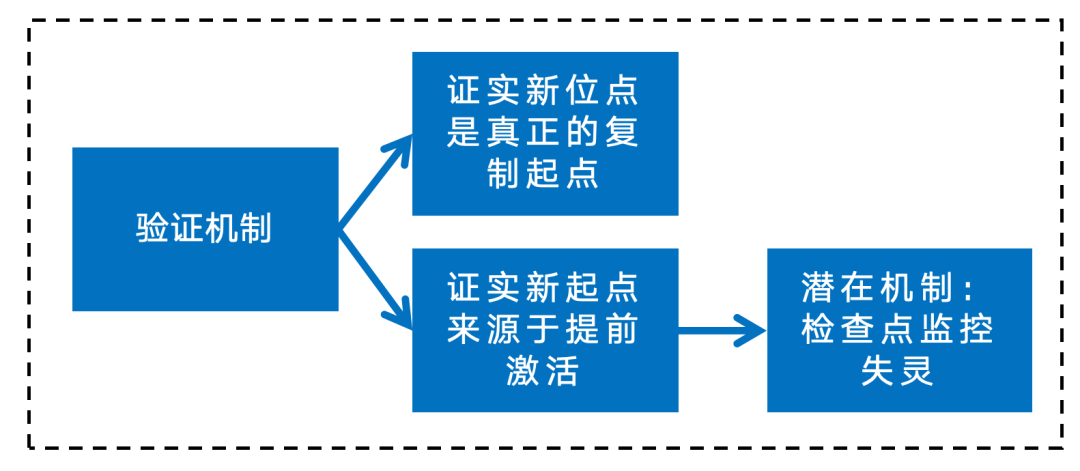
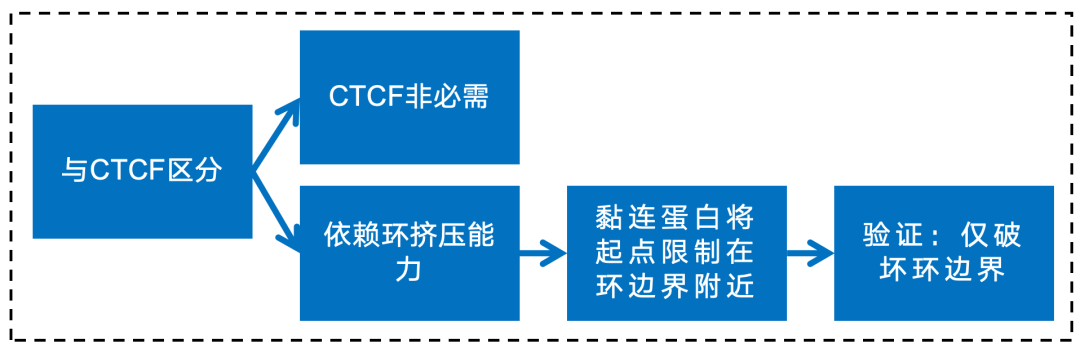
英文题目:Cohesin maintains replication timing to suppress DNA damage on cancer genes 中文题目:黏连蛋白通过维持复制时序来抑制癌基因上的DNA损伤 期刊:Nature Genetics(IF:29) 单位:北京大学生命科学学院 发表时间:2023年8月 摘要: 研究发现,黏连蛋白功能丧失突变在肿瘤中频繁出现,但其在肿瘤发生中的作用机制尚不明确。本研究发现,黏连蛋白核心亚基RAD21的缺失会导致全基因组范围内大规模DNA断裂,并形成147个染色体易位热点基因——这些基因在多种癌症中与黏连蛋白共突变。DNA损伤增加与RAD21缺失引起的转录改变及染色质环锚点破坏无关,但损伤诱导的染色体易位与冈崎片段的不对称分布重合,提示RAD21缺失会引发复制压力,表现为复制速度减慢和停滞复制叉增加。机制上,RAD21缺失导致900多个休眠复制源过早启动,使约30%的人类基因组出现复制时相提前。相应地,大多数易位热点基因位于复制时相改变区域。由此得出结论:黏连蛋白功能失调通过诱发过度DNA复制起始导致复制压力,进而产生可能促进肿瘤发生的大规模DNA损伤。 思维导图: 研究背景: 1.背景知识: (1)黏连蛋白的结构与功能:它是一个环状复合体,核心功能包括姐妹染色单体黏附、染色体分离,并与CTCF合作形成染色质环以调控基因表达。 (2)临床关联:黏连蛋白亚基在多种癌症中发生频繁突变,与肿瘤发生相关。 2.现存问题/知识空白: (1)黏连蛋白突变导致癌症的具体机制不明。 (2)传统的认知(如染色体分离错误、转录调控紊乱)可能不是其主要致癌原因。 (3)黏连蛋白在DNA复制中的作用,特别是对复制叉进程和复制时序的影响,是尚未深入探索的关键领域。 3.本研究的目标与核心发现: (1)研究手段:使用先进的降解子系统快速去除黏连蛋白核心亚基RAD21。 (2)研究目标:旨在解决“黏连蛋白突变如何致癌”这一难题,并通过实验提出,其核心机制在于黏连蛋白通过调控DNA复制时序来维持基因组稳定性,而非主要通过影响染色体分离或全局转录。 主要结果: 1.RAD21缺失引起全基因组DNA断裂 黏连蛋白亚基在白血病、子宫内膜癌、黑色素瘤等多种癌症类型中频繁发生突变(附图1a)。通过使用SIFT(耐受性排序)工具分析,研究者发现每个黏连蛋白亚基中超过52%的癌症突变被预测为功能丧失型,包括移码突变、截短突变、有害替换、插入和缺失(附图1b)。其中,RAD21亚基的功能丧失突变频率最高,达到65.3%(附图1b)。为研究黏连蛋白功能障碍,研究者在K562细胞中采用微型生长素诱导降解系统实现RAD21的快速降解(图1a及附图1c,d)。具体策略为:将OsTIR1基因敲入AAVS1位点,同时在RAD21基因的3'端引入mAID-mClover序列(图1a及附图1c,命名为RAD21-mAC),并成功获得两个纯合克隆#1和#4(图1a及附图1c,d)。经吲哚-3-乙酸处理后6小时或24小时,分别有超过20%或24%的RAD21-mAC细胞中RAD21表达水平与未处理组相当(图1a及附图1d)。为排除IAA耐药细胞的干扰,研究者通过流式分选技术分离出mClover荧光不可测的RAD21缺失细胞用于后续分析。分选后的IAA处理组RAD21-mAC细胞中RAD21蛋白完全不可检测,染色质结合的SMC1蛋白量也显著减少,表明黏连蛋白复合体已解离(图1a)。值得注意的是,RAD21降解后染色质上的CTCF蛋白量保持稳定(图1a)。此外,RAD21缺失细胞中γH2A.X焦点数量显著增加,提示RAD21缺陷在人类细胞中引发了大量DNA断裂(图1b,c及附图1e)。 在小鼠胚胎干细胞中,研究者采用与K562细胞相似的策略构建了Rad21-mAC细胞系(附图2a,b)。IAA处理3小时后,超过80%的Rad21-mAC细胞中RAD21-mAC蛋白降至不可检测水平(附图2a,b)。通过端粒荧光原位杂交技术对染色体单体断裂进行定量分析发现(附图2c,d),Rad21-mAC mES细胞中RAD21缺失导致姐妹染色单体黏连缺陷(附图2e),且染色体断裂发生率比野生型细胞高出约三倍(附图2c,d),这表明RAD21在小鼠胚胎干细胞中同样具有维持基因组稳定性的功能。 2.RAD21缺失导致癌症相关基因发生反复断裂 为全面表征黏连蛋白缺失引发的DNA双链断裂,研究者运用已建立的引物延伸测序技术对K562细胞进行全基因组DSB定量分析。该技术通过将CRISPR-Cas9诱导的“诱饵”DSB位点与全基因组“猎物”DSB位点形成的易位连接体进行捕获(图1d),经单轮引物延伸及携带独特分子标识的桥接头连接,可有效排除PCR重复并实现与诱饵DSB形成染色体易位的全基因组DSB定量检测。研究者在野生型及RAD21-mAC克隆#1/#4的K562细胞中,针对MYC或TP53诱饵位点构建了有无RAD21存在条件下的三组生物学重复样本(图1d)。检测发现RAD21缺失细胞中易位断裂位点显著增加,特别是在含诱饵位点的染色体上。全基因组DSB水平较IAA处理的野生型细胞及未处理RAD21-mAC细胞升高3-5倍(图1e及附图2f,g)。通过外源表达野生型RAD21蛋白可显著逆转IAA处理RAD21-mAC细胞中升高的DSB水平(图1f及附图2h),而两种白血病来源的RAD21突变体(已知可增强干细胞特性)则无法挽救此表型(图1f及附图2h)。鉴于癌细胞中黏连蛋白突变常呈杂合状态,研究者使用短发夹RNA在K562细胞中部分敲低RAD21表达(附图2i),每种shRNA均导致靶细胞中RAD21蛋白部分缺失(50-80%),并通过PEM-seq检测到全基因组DSB水平显著上升,且DSB增幅与RAD21缺失程度呈正相关(附图2j,k)。这些数据共同证明RAD21完全或部分缺失均会引发全基因组DNA断裂。 通过设定错误发现率<0.01的统计学标准,研究者在RAD21缺失细胞中鉴定出显著聚集的易位断裂簇。在MYC和TP53诱饵位点分别检测到127个和158个易位簇(附图3a),最终确定147个易位热点基因,其中76个基因在双诱饵系统中共同出现(图2a,b、附图3b,c及附表1)。超过三分之一的热点基因在人类蛋白质图谱数据库中被注释为癌症生物标志物或疾病相关基因,包括STAG2、KMT2C、RUNX1和BCR等(图2b、附图3c及附表1)。后续通过END-seq技术检测未连接DSB发现,RAD21缺失时全基因组DSB数量较IAA处理的野生型细胞增加1.8-2.3倍(附图3d),与PEM-seq结果吻合。特别值得注意的是,RAD21缺失后PEM-seq鉴定热点基因上的DNA断裂增加2.1-2.9倍(图2c及附图3e),且这些基因在癌症中更易形成异常融合(附图3f),提示其基因组脆弱性特征。 为探究黏连蛋白突变是否通过提升热点基因突变率驱动肿瘤发生,研究者对五种与RAD21功能障碍相关的癌症类型(包括RAD21高频突变的子宫内膜癌、黑色素瘤,以及已报道与RAD21功能障碍相关的结直肠癌、非小细胞肺癌和胶质瘤)进行共突变分析。结果显示,在携带黏连蛋白突变的患者中,77.6-98.3%的热点基因突变频率显著高于无黏连蛋白突变群体(图2d及附图3g)。以黑色素瘤为例,COL5A1基因在总体患者中突变率为8.8%,而在黏连蛋白突变亚群中升至29.6%,非突变亚群中仅为6.7%(图2d)。进一步发现147个热点基因中有82个在五种癌症中均与黏连蛋白显著共突变(图2d-f及附图3g),其中STAG1和STAG2不仅被鉴定为热点基因,还在癌症中呈现高频突变且与其他黏连蛋白亚基显著共突变(图2f)。综上,这些数据表明RAD21缺失会导致癌症相关基因发生反复性DNA断裂。 3.染色质环或转录改变并非DSBs主因 黏连蛋白通过将DNA外推至染色质环锚定点,对染色质环的形成至关重要。经比较发现,IAA处理与未处理的野生型细胞中黏连蛋白全基因组分布基本一致(附图4a左图),但在IAA处理的RAD21-mAC细胞中,染色质结合的RAD21蛋白被完全清除(图3a,b、附图4a右图及附图4b)。为验证RAD21缺失对染色质环的影响,研究者在MYC基因座建立了定量染色质构象捕获测序技术(图3a)。通过将引物置于MYC近端上游CTCF结合元件,并采用PEM-seq的定量策略,发现RAD21缺失细胞中环锚点间的染色质相互作用完全消失(图3a)。然而,环锚点区域的易位频率在RAD21缺失条件下仍保持稳定(图3c),这表明染色质环边界稳定性的改变可能并非黏连蛋白功能障碍导致DSB形成的主要机制。 既往研究表明黏连蛋白缺失仅引起HCT116细胞中轻微的转录水平改变。通过精度核运行测序技术分析新生RNA发现,RAD21完全缺失仅导致K562细胞发生有限的转录改变——在147个热点基因中,有140个基因(如SMYD3和AGAP1)在RAD21存在与否条件下转录水平保持稳定(图3d,e及附图4c)。与之相符的是,作为DSB形成常见区域的转录起始位点周边,其易位频率也未出现显著变化(图3f)。这些数据共同表明,RAD21缺失仅诱导约1.0%的基因发生转录改变,且这些改变与RAD21缺失条件下的易位热点分布无显著关联。 4.RAD21缺失诱导的DSBs与DNA复制过程吻合 为探究DNA复制是否参与RAD21缺失后DSB的形成,研究者通过冈崎片段测序技术对DNA复制过程中沃森链(W)与克里克链(C)的复制叉方向进行图谱分析(图4a,b)。引人注目的是,RAD21缺失细胞中的易位连接点在诱饵切割位点周围呈不对称分布。对已发表的OK-seq信号数据重新分析发现,这些易位连接点的分布模式与OK-seq图谱高度吻合(图4a及附图5a)。进一步观察发现,易位连接点倾向于出现在OK-seq沃森峰区域的沃森链上,而在克里克峰区域则呈现相反规律(图4b,c)。统计学分析显示易位连接点与OK-seq信号呈正相关,皮尔逊相关系数大于0.5(附图5b)。此外,RAD21缺失后全基因组范围的沃森/克里克峰区域出现轻微增多的易位连接点积累(附图5c)。值得注意的是,在复制叉方向转换区域,易位连接点呈现与OK-seq信号方向一致的转换模式(图4d)。这些数据共同提示,RAD21缺失诱导的DSB可能与DNA复制过程相关(附图5d)。 随后研究者采用低剂量羟基脲或阿非迪霉素诱导复制压力,并通过PEM-seq检测K562细胞中的DNA断裂。结果显示,两种药物诱导的DNA断裂均呈现与OK-seq图谱相似的转换模式(图4e及附图5e),而主要诱导转录相关DSB的依托泊苷处理则未显示此相关性(图4e)。通过DNA分子梳技术分析DNA复制动态发现(图4f),RAD21缺失导致复制速度显著降低,同时停滞复制叉频率增加(图4g,h)。这些结果表明RAD21缺失可能通过诱发复制压力导致DNA断裂。 5.RAD21缺失细胞中紊乱的DNA复制时序 黏连蛋白在调控DNA复制叉进程中的作用尚存争议,且其功能障碍对复制时相维持的整体影响仍未明晰。为探究RAD21缺失诱导复制压力的根源,研究者通过复制时相分析展开研究。将生长中的细胞用EdU标记15分钟后,根据DNA含量分选为六个组分,采用核苷类似物掺入位点测序技术构建测序文库,并运用RepFind流程进行数据处理。以S50值作为复制时相指标,该值代表基因组区域在50%细胞中完成DNA复制的时刻,其数值范围从0(早复制)到1(晚复制)。 研究发现RAD21缺失细胞在某些基因组区域呈现更早的复制时相。通过野生型细胞两个重复样本的复制时相相关性图谱确定95%分位数区间,作为时相未改变区域的评估标准。结果显示在IAA处理6小时或24小时的RAD21-mAC细胞中,分别有约7.9%和30.1%的基因组区域出现显著提前的复制时相。值得注意的是,IAA处理的小鼠胚胎干细胞也显示约13.4%的基因组区域在RAD21缺失后复制时相提前,且RAD21缺失后更多基因组区域在S期早期完成复制。 研究者进一步分析了黏连蛋白功能障碍下复制时相与DSB的关联。发现在RAD21缺失24小时后,超过82%的易位热点基因落入早期复制阶段(S50<0.5),且与野生型细胞相比,大多数热点基因在RAD21缺失细胞中呈现提前而非延迟的复制时相。例如MYC诱饵捕获的PTPRN2和STAG2基因,以及TP53诱饵捕获的CCNY和STAG1基因均表现出此特征。这些数据共同提示,RAD21缺失引起的基因组不稳定性可能源于过早的额外复制事件。 6.RAD21缺失细胞中异常的DNA复制起始 异常的复制起始过早可能导致额外早期复制现象。通过对早期S期阻滞细胞的新生DNA进行标记,发现两个RAD21-mAC克隆在RAD21缺失后,早期复制灶数量均增加至1.5倍以上(图6a,b及附图7a)。为精确鉴定RAD21缺失细胞中真正的早期复制起始区,研究者采用NAIL-seq技术进行分析:将细胞同步于G1期,IAA处理6小时后释放至G1/S转换期进行EdU-BrdU序贯标记(附图7b)。尽管阻滞的RAD21缺失细胞进入S期的进程较野生型缓慢,但仍能正常进入复制阶段。为捕获早期DNA复制起始事件,分别在释放后4小时和3小时收集RAD21缺失细胞与野生型细胞样本。值得注意的是,在释放前染色质上的复制起始识别复合体及微型染色体维持复合体含量保持稳定(附图7d)。 各细胞类型中三个生物学重复的早期复制信号均呈现高度可重复性,因此将各克隆的合并信号用于后续分析(附图7e)。研究者在IAA处理的野生型细胞中鉴定出2603个早期复制起始区,与既往研究结果一致。在RAD21-mAC克隆#1和#4中分别鉴定出3715和3739个早期复制起始区,且超过90%的起始区为两克隆共有。引人注目的是,RAD21缺失的#1和#4细胞中分别新出现997和985个早期复制起始区,约占各克隆总起始区数量的25%。通过比较野生型与RAD21缺失细胞共有起始区的复制信号强度,研究者鉴定出“增强型”(#1克隆514个,#4克隆494个)、“持平型”(#1克隆1939个,#4克隆1957个)和“减弱型”(#1克隆265个,#4克隆303个)三类起始区,分别对应RAD21缺失后早期复制信号上调、等效或下调的情况。此外,在野生型细胞中存在226/222个“消失型”起始区未出现在RAD21缺失细胞中。但由于这些起始区在野生型细胞中的总复制信号本已较弱,且在RAD21缺失细胞中仅轻微降低,故将其归入“减弱型”起始区进行后续分析(图6d-f)。总体而言,RAD21缺失后仅约半数的早期复制起始区保持相当的复制信号强度。 7.黏连蛋白耗竭后休眠复制起点过早激活 研究者在RAD21缺失细胞中进一步发现,复制起始相关因子(包括ORC、MCM复合物及组蛋白变体H2A.Z)在新出现的早期复制起始区富集,这与先前鉴定的其他类型起始区特征相似(图6c,g)。值得注意的是,包括新型起始区在内的所有四类起始区均缺乏转录活性,这与既往研究结果一致。此外,新起始区中心检测到指示复制起始的OK-seq信号急剧转换特征(图6g)。这些证据共同表明,RAD21缺失细胞中的新型早期复制起始区具备DNA复制源的典型特征。 通过将早期复制起始区定位至复制时间轴进行分析,发现"持平型"起始区在野生型和RAD21缺失细胞中的复制时相基本一致,“减弱型”起始区在RAD21缺失细胞中呈现轻微延迟。与之形成鲜明对比的是,“增强型”和“新型”起始区所在基因组区域在RAD21降解后均表现出显著的复制时相提前现象(图6h)。这一结果提示,RAD21缺失细胞中的新型起始区可能源于休眠复制源的过早激活。由于S期内检查点通常抑制休眠源的活化,而RAD21缺失细胞中两个关键检查点激酶(CHK1和CHK2)均未充分磷酸化,从而解除了对休眠复制源的抑制。 同样在小鼠胚胎干细胞中也观察到类似现象:Rad21缺失细胞比野生型细胞激活更多复制起始区,其中15.6%的早期复制峰为新出现区域,这些新型起始区导致约13.4%的基因组区域复制时相提前。这些发现共同表明,RAD21通过抑制休眠复制源的过早利用来维持正常的复制时相程序。 8.黏连蛋白在染色质环内抑制早期复制起点激活 鉴于黏连蛋白与CTCF在染色质环锚定中均发挥关键作用,研究者进一步探究CTCF是否参与调控新型早期复制起始区的过早激活。通过构建CTCF-mAID-mClover融合蛋白细胞系,并进行NAIL-seq EdU/HU实验分析,发现CTCF缺失细胞中已鉴定起始区的复制信号图谱与野生型细胞基本相似,仅信号强度略有降低。值得注意的是,与野生型细胞相同,CTCF缺失细胞在新型起始区仅表现出极微弱的复制信号,这表明CTCF并非抑制休眠复制源激活的必要条件。因此,黏连蛋白不依赖于CTCF的染色质环挤出功能可能在协调DNA复制起始过程中发挥关键作用。 哺乳动物细胞中复制源的激活需要MCM双六聚体的装载与活化。鉴于黏连蛋白可与MCM复合体相互作用,研究者探究了黏连蛋白是否通过重新分布MCM来调控复制源激活。在野生型细胞中,K562和小鼠胚胎干细胞均显示早期复制倾向于发生在靠近染色质环边界的区域(以MYC基因座为例)。统计分析发现,新型和增强型起始区与环边界的平均距离分别达到45.0 kb和39.0 kb,显著大于野生型细胞所有起始区的平均距离(30.5 kb)。通过在G1期阻滞细胞中进行MCM5染色质免疫沉淀测序分析(该时期正发生复制源许可),发现黏连蛋白缺失细胞中MCM在染色质环结构域内部区域的积累显著高于野生型细胞,这一分布特征与新型起始区的定位相吻合。这些数据共同提示,黏连蛋白可能通过抑制环边界远端休眠复制源的异常激活来维持复制程序的稳定性。 为验证上述发现,研究者通过CRISPR-Cas9技术敲除MYC环边界的CTCF结合元件,构建了CBE+/- K562细胞系。定量染色质构象捕获测序显示,部分CBE缺失削弱了该区域与下游环锚点(特别是PVT1基因下游CBE)的染色质相互作用。与此同时,在相互作用减弱的区域检测到更多早期复制起始信号,提示该区域复制时相提前。通过在下游558 kb处设置诱饵DSB进行PEM-seq检测,发现CBE+/-细胞中相互作用减弱区域的DSB数量达到野生型细胞的两倍。与此一致,END-seq技术在同一区域也检测到CBE缺失后DSB水平升高3.7倍。 讨论 黏连蛋白突变已被证实是白血病、结直肠癌等多种肿瘤的驱动因素。不同癌症类型中反复出现的黏连蛋白突变现象提示,其功能丧失是以全局性而非位点特异性的方式破坏基因组完整性。因此,阐明黏连蛋白在肿瘤发生中的潜在机制具有日益重要的临床意义。本研究通过采用蛋白降解系统特异性去除黏连蛋白核心亚基RAD21,系统揭示了其在DNA复制与基因组稳定性维护中的关键作用,这为理解黏连蛋白功能障碍如何促进肿瘤进展提供了新的机制视角。 DNA复制时相是确保基因组精确复制的稳健保守程序,其改变与常见脆弱位点相关,并在某些肿瘤中普遍存在。RAD21缺失导致休眠复制源在S期早期被激活,进而可能扰乱正常复制时相。相较于野生型细胞,RAD21缺失的K562细胞中早期复制起始区数量增加30%,这与既往研究中RAD21缺失导致更多早期复制域的现象一致。尽管早期阶段有更多复制源被激活,但黏连蛋白缺失细胞的复制速度反而减慢,这与通过消除SMC3亚基乙酰化导致复制叉进程减慢的研究结果相符。RAD21缺失可能通过多种机制诱导过度的DNA复制起始事件,从而威胁基因组完整性:其一,复制起始过程本身可能偶然引发DSB,已有证据显示小鼠B细胞中早期复制与DSB及DNA修复因子存在共定位;其二,过量复制叉会消耗更多复制因子,当限制性复制因子耗竭时将引发严重复制压力;其三,过量复制叉与转录过程的碰撞以及复制终止事件增加也可能导致DSB形成。 在RAD21缺失的K562细胞中,全基因组DSB水平升高3-5倍。综合来看,DNA复制而非转录或染色质环破坏更可能是导致大规模DNA损伤的主要因素。在此背景下,PEM-seq检测到的DSB与复制叉中冈崎片段呈正相关。值得注意的是,通过改变特定基因座的复制时相可降低激活诱导胞苷脱氨酶依赖的致癌易位发生率,这提示癌基因在复制压力下的脆弱性。黏连蛋白还通过ATM损伤应答通路参与DNA修复,但其缺失诱导的DSB增幅高于ATM缺陷B细胞中观察到的两倍增幅,这表明多数易位事件不依赖ATM通路,尽管黏连蛋白缺失也可能通过削弱DNA修复能力加剧基因组不稳定性。 研究未发现CTCF参与抑制休眠复制源的激活,提示黏连蛋白不依赖CTCF的染色质环挤出功能可能负责复制调控。鉴于MCM双六聚体在HeLa细胞中与黏连蛋白共定位,研究者提出黏连蛋白可能在染色质环挤出过程中沿环结构重新分布未激活的MCM复合体。相应地,RAD21缺失后MCM复合体在染色质环结构域内部区域呈现更高积累。相反,在导致RNA聚合酶II暂停降解的α-鹅膏蕈碱处理后,MCM5在环结构域中的分布几乎不变,这可能是通过重新分布MCM复合体以确保非转录区域的复制起始。总之,MCM分布可能受染色质环挤出与转录过程共同调控,导致DNA复制起始聚集在靠近环边界的非转录区域。缺乏黏连蛋白时,MCM分布仍可能受转录调控,因为复制起始信号仍局限于非转录区域。因此,MCM六聚体可能同时存在于环结构域的早期复制源与休眠复制源中,这使得黏连蛋白缺失时远离环边界的区域仍保留诱导额外DNA复制起始的潜力。此外,HCT116细胞中黏连蛋白敲低导致环边界处某些高效复制起始区发生去定位化的现象也支持研究者的假说。然而,黏连蛋白与MCM相互作用的分子机制仍有待进一步探索。 END 阅读最新文献,紧跟前沿进展,这是一名研究者必须具备的习惯和要求。我们华西医院耳鼻咽喉头颈外科的硕士、博士研究生和博士后们自2019年以来,每周开展一次文献泛读和文献精读分享会,至今已累计开展了200多次。2023年9月13日开始,本科室陆续将其进行整理,同步推出在线前沿速递和文献解读板块。通过这种学习和分享的方式,使汇报者和大家都能对近期权威期刊发表的高质量研究有所了解,同时也是学习其他优秀研究者思路、方法和理论的良好手段。希望通过这种形式,把科内的分享扩大到所有的读者,一起学习,共同进步! 华西医院耳鼻咽喉头颈外科 2023年9月13日