



精读分享│:靶向TBK1以克服癌症免疫治疗的耐药性
英文题目:Targeting TBK1 to overcome resistance to cancer immunotherapy
中文题目:靶向TBK1以克服癌症免疫治疗的耐药性
期刊:Nature(IF=50.5)

Abstract
Despite the success of PD-1 blockade in melanoma and other cancers, effective treatment strategies to overcome resistance to cancer immunotherapy are lacking. Here we identify the innate immune kinase TANK-binding kinase 1 (TBK1) as a candidate immune-evasion gene in a pooled genetic screen. Using a suite of genetic and pharmacological tools across multiple experimental model systems, we confirm a role for TBK1 as an immune-evasion gene. Targeting TBK1 enhances responses to PD-1 blockade by decreasing the cytotoxicity threshold to effector cytokines (TNF and IFNγ). TBK1 inhibition in combination with PD-1 blockade also demonstrated efficacy using patient-derived tumour models, with concordant findings in matched patient-derived organotypic tumour spheroids and matched patient-derived organoids. Tumour cells lacking TBK1 are primed to undergo RIPK- and caspase-dependent cell death in response to TNF and IFNγ in a JAK–STAT-dependent manner. Taken together, our results demonstrate that targeting TBK1 is an effective strategy to overcome resistance to cancer immunotherapy.
摘要
尽管PD-1阻断疗法在黑色素瘤等癌症治疗中取得显著成效,但针对癌症免疫疗法耐药性的有效治疗策略仍属空白。该研究通过综合遗传筛选,将固有免疫激酶TANK结合激酶1(TBK1)鉴定为潜在的免疫逃逸相关基因。研究者运用多套遗传学工具与药理学手段,在多种实验模型体系中验证了TBK1作为免疫逃逸基因的作用。靶向TBK1可通过降低效应细胞因子(TNF与IFNγ)的细胞毒性阈值,显著增强PD-1阻断疗法的应答效果。基于患者来源肿瘤模型的实验显示,TBK1抑制剂与PD-1阻断剂的联合疗法具有显著疗效,这一发现在匹配的患者来源类器官肿瘤球模型及类器官模型中均获得一致性验证。机制研究表明,TBK1缺失的肿瘤细胞在JAK-STAT信号通路介导下,会启动对TNF和IFNγ的敏感性,进而通过RIPK及半胱天冬酶依赖性途径发生细胞死亡。综上所述,该研究证实靶向TBK1是克服癌症免疫疗法耐药性的有效策略。
研究背景:
尽管免疫检查点阻断(ICB)疗法革新了晚期黑色素瘤等癌症的治疗格局,但克服耐药性仍是核心挑战。目前对于先天或获得性ICB耐药患者尚无获批疗法。为突破原发性耐药,多项评估新型免疫调节剂联合抗PD-1/PD-L1疗法的临床试验正在进行。值得注意的是,近期两项III期随机对照临床试验显示,颇具前景的联合方案均未较PD-1单药治疗展现出生存优势,这促使学界重新聚焦联合策略的临床前及早期临床开发。
基于CRISPR-Cas9基因组编辑的功能缺失性遗传筛选作为无偏靶点发现手段,已成功鉴定出多个增强抗肿瘤免疫应答的靶点。多项体内外CRISPR-Cas9筛选研究揭示了肿瘤内在的免疫治疗耐药驱动因子,但这些发现的临床应用仍显局限。
TANK结合激酶1(TBK1)作为多功能丝氨酸/苏氨酸激酶,在协调抗病毒等先天免疫应答中具有明确作用。该激酶通过整合模式识别受体与胞浆核酸传感器的上游信号,调控干扰素调节因子3(IRF3)活化,进而诱导I型干扰素(IFNα/β)及干扰素刺激基因(ISGs)表达。鉴于激活胞浆核酸传感通路是"免疫冷肿瘤"促炎化的潜在策略,TBK1被鉴定为免疫逃逸候选基因且其信号阻断可增强ICB疗效的发现显得尤为意外。这些看似矛盾的证据使得TBK1调控癌症免疫治疗敏感性的确切机制亟待阐明。
该研究通过经典小鼠肿瘤模型与患者来源模型证实:TBK1基因缺失可增敏肿瘤免疫攻击,其药理抑制能克服PD-1阻断耐药。靶向TBK1可通过降低免疫细胞效应细胞因子的细胞毒性阈值,使耐药肿瘤重获ICB敏感性。
研究方法和思路:
研究结果:
(1)TBK1的缺失使肿瘤对免疫检查点抑制治疗(ICB)更敏感
在前期开展的体内CRISPR筛选中,研究者观察到:经PD-1阻断治疗后,免疫健全小鼠的B16黑色素瘤肿瘤中靶向Tbk1的单向导RNA(sgRNA)显著减少(图1a),提示TBK1缺失的肿瘤细胞更易被免疫系统控制。值得注意的是,靶向同源先天免疫信号激酶IKKε(Ikbke)的sgRNA并未出现富集现象,表明该效应具有TBK1特异性。
为验证Tbk1缺失能否增强PD-1阻断疗效,研究者采用两种不同sgRNA通过CRISPR-Cas9基因敲除技术构建了Tbk1缺陷型B16小鼠黑色素瘤细胞株,并通过蛋白印迹证实了TBK1蛋白表达的缺失。在体外培养条件下及植入免疫缺陷型NSG小鼠体内时,Tbk1敲除细胞与对照细胞的增殖速率相当(图1b)。
在免疫健全野生型小鼠模型中,Tbk1敲除与对照B16肿瘤的生长曲线及生存期基本一致;而接受抗PD-1治疗后,与对照组相比,携带Tbk1敲除肿瘤的小鼠展现出更显著的肿瘤消退和生存优势(图1c)。这些结果证实:Tbk1缺失的B16肿瘤在体内生长特性正常,但对PD-1阻断免疫治疗具有更高敏感性。
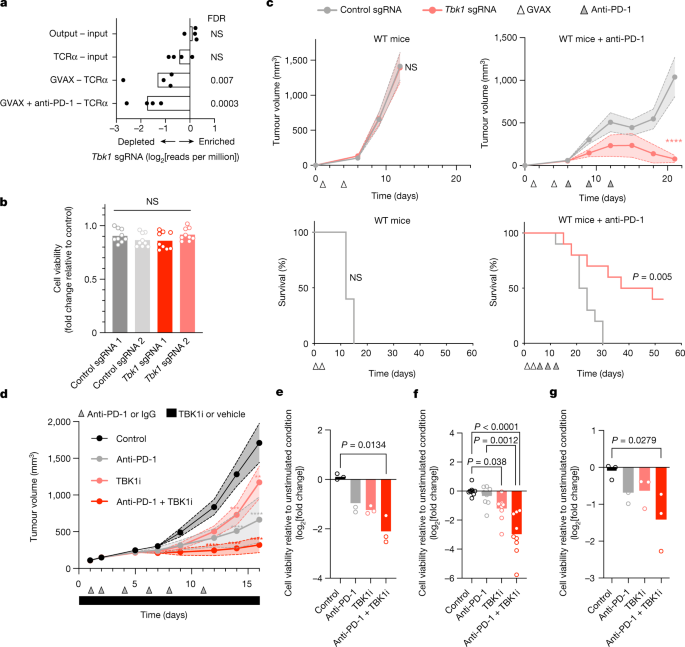
图1:TBK1缺失使肿瘤对PD-1阻断治疗敏感
(2)抑制TBK1可增强对免疫检查点抑制治疗的响应
TBK1在固有免疫感应过程中具有重要作用,目前其抑制剂正被评估用于自身免疫性疾病及炎症性疾病的治疗。这引发一个重要问题:系统性抑制TBK1可能削弱炎症反应,从而无法复现肿瘤特异性TBK1缺失所介导的增敏效应。为此,研究者探究了TBK1药理抑制能否模拟TBK1缺失B16肿瘤的表型特征。
研究者采用表达模型抗原卵清蛋白的B16肿瘤(B16-ova)的荷瘤野生型小鼠模型,分别给予IgG对照、抗PD-1单药、或联合使用抗PD-1与已报道的小分子TBK1抑制剂(TBK1i)进行治疗。结果显示,相较于单药组和对照组,抗PD-1联合TBK1i治疗组小鼠的肿瘤控制显著改善(图1d),且耐受性良好,未观察到毒性反应或体重下降。通过B16-ova荷瘤小鼠来源的类器官肿瘤球(MDOTS)离体实验证实,联合治疗组对抗PD-1的敏感性显著增强(图1e)。在CT26 MDOTS模型(对PD-1阻断单药或联合治疗部分敏感)中,抗CD8α抗体处理实验表明,CD8+T细胞活性是抗PD-1与TBK1i协同效应的必要条件。此外,在抗PD-1耐药的D4M.3A(Braf突变/Pten缺失)肿瘤来源的MDOTS中,TBK1i成功克服了原发性(内在)耐药(图1f);而在体内已产生获得性(继发)PD-1阻断耐药的B16-ova MDOTS模型中,同样观察到TBK1i的增敏效应(图1g)。在MC38(敏感型)和MB49(部分敏感型)同源小鼠肿瘤模型中,TBK1i联合PD-L1阻断也展现出更强的体内肿瘤控制效果。这些发现共同证明,TBK1抑制剂联合抗PD-1治疗在原发性与继发性PD-1阻断耐药的小鼠肿瘤模型中均具有显著活性。
(3)TBK1抑制剂在PDOTS中增强了对ICB的响应
为探究TBK1抑制策略对人类癌症ICB原发性或获得性耐药的克服作用,研究者采用患者来源类器官肿瘤球(PDOTS)进行了离体药效评估(图2a)。研究纳入了30例PDOTS样本,包括皮肤黑色素瘤(15例)、非皮肤黑色素瘤(2例)及其他癌种(13例)。离体培养结果显示,相较于PD-1单药阻断(16.6%应答率),TBK1抑制剂单药(30%应答率)及其与PD-1阻断联用(40%应答率)能显著抑制肿瘤生长。IgG4同型抗体对照组未显现效应,与既往报道一致。
值得注意的是,来自免疫治疗耐药转移性皮肤黑色素瘤患者的PDOTS对TBK1i联合抗PD-1治疗敏感,而对PD-1单药或联合CTLA-4阻断均无应答(图2c,d)。在其他癌种中亦观察到TBK1i联合PD-1阻断的显著疗效,尤其对于存在微卫星不稳定性(MSI)证据的结直肠癌(图2e,f)。这些数据通过临床ICB耐药患者来源模型,证实了TBK1抑制剂联合PD-1阻断的治疗潜力。
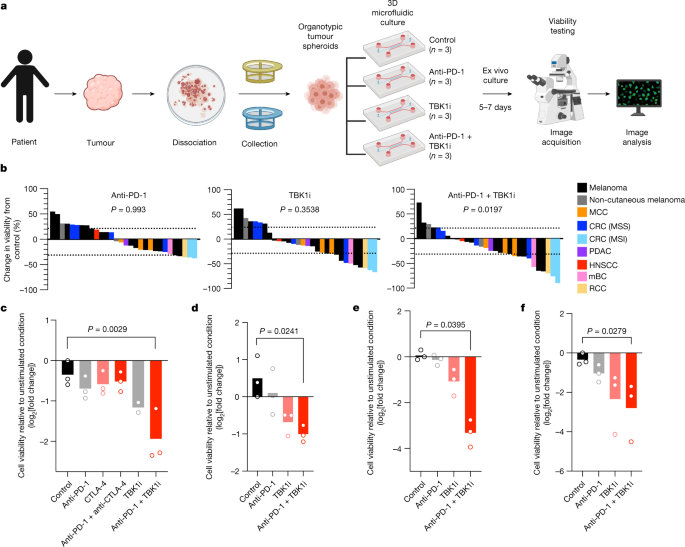
图2:TBK1抑制剂通过PDOTS模型增强对PD-1阻断的敏感性
(4)TBK1抑制剂与肿瘤免疫环境
TBK1及其同源激酶IKKε(由IKBKE编码)在人类黑色素瘤的淋巴细胞和髓系细胞中广泛表达。近期研究揭示TBK1/IKKε在调控T细胞、B细胞、树突状细胞及巨噬细胞等多种免疫细胞活性中发挥着关键作用。为解析TBK1抑制对肿瘤免疫微环境的影响,研究者对来自接受抗 PD-1、TBK1i 或抗 PD-1 加 TBK1i 治疗的B16-ova荷瘤小鼠的CD45+细胞(n=53,637)进行了单细胞RNA测序(scRNA-seq)分析,并与使用同型对照(IgG)治疗的情况进行比较(图3a)。通过整合各治疗组数据进行聚类分析,创建跨条件稳定的聚类集,然后量化了不同条件间群体相对丰度的变化(图3b)。
结果显示,抗PD-1治疗组T细胞与自然杀伤(NK)细胞群体显著扩增(图3b),终末耗竭/效应CD8+T细胞比例增加;而TBK1i治疗组(无论是否联合抗PD-1)则富集早期耗竭/效应CD8+T细胞,终末耗竭亚群减少。体外实验证实,TBK1i处理的小鼠脾源性T淋巴细胞其细胞因子(TNF、IL-2、IFNγ)分泌增强,与效应功能增强一致。
在用TBKi治疗的或同时用抗PD-1治疗的肿瘤小鼠中观察到显著增加的髓系细胞(图3b)。进一步亚群分析显示,促炎性巨噬细胞(如M1型)增多,而髓系来源抑制细胞(MDSCs)等免疫抑制性群体减少(图3c-e)。基因集富集分析表明,TBK1i治疗与TNF-NF-κB信号及炎症相关通路激活显著相关(图3f,g)。髓系细胞亚群中TNF(Tnf)与IL-1α(Il1a)表达增强,在TBK1i治疗组尤为显著(图3h,i)。TBK1i预处理可增强骨髓源性巨噬细胞在LPS和IFNγ刺激下的Tnf/Il1a的表达(图3j),证实TBK1抑制直接调控髓系细胞炎症反应。这些发现表明TBK1i具有肿瘤外效应,在单独使用或与抗PD-1治疗联用时,均能显著重塑髓系细胞亚群,同时TBK1i足以增强肿瘤微环境中炎症细胞因子(如IFNγ和TNF)的表达。。
研究者进一步探究肿瘤特异性TBK1缺失是否影响免疫微环境。对植入野生型小鼠并接受抗 PD-1 治疗的对照组和 Tbk1 敲除型 B16 肿瘤的肿瘤浸润免疫细胞进行流式细胞术分析发现,CD8+/CD4+T细胞、GZMB+CD8+T细胞、FOXP3+调节性T细胞、NK细胞及F4/80+髓系细胞均无显著差异。使用scRNA-seq分析抗PD-1治疗后的CD45+细胞(n=31,810)显示,Tbk1缺失仅引起CD8+T细胞与M1样巨噬细胞轻度增加。TBK1表达谱分析证实其在巨噬细胞、MDSCs和CD8+T细胞中表达最高,且Tbk1缺失肿瘤细胞中IKKε表达完整保留。这些结果说明TBK1缺失肿瘤对抗PD-1治疗的增敏效应不依赖于免疫区室重塑,支持TBK1作为"肿瘤内在"免疫逃逸基因的功能定位。
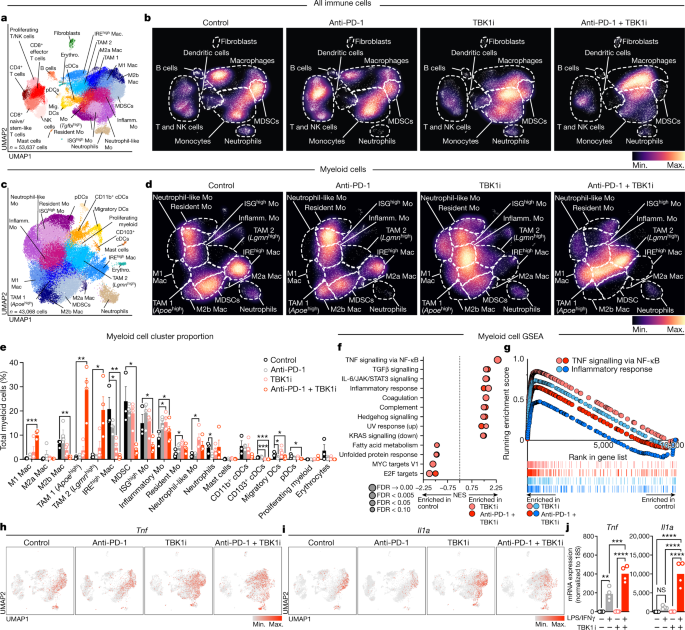
图3:TBK1抑制剂重塑肿瘤免疫微环境
(5)TBK1的缺失增强了肿瘤对TNF和IFNγ的敏感性
IFNγ与TNF是介导抗肿瘤免疫应答的关键效应细胞因子,但IFN(干扰素)和TNF信号通路相关基因有助于免疫逃逸。在203例转移性黑色素瘤患者队列中,虽然ICB治疗6周时应答与非应答患者均出现循环TNF和IFNγ水平升高,但非应答者在治疗6个月后仍维持高细胞因子水平(图4a,b)。黑色素瘤患者的scRNA-seq数据证实,非应答肿瘤中IFNG和TNF表达显著高于应答组(图4c)。其中IFNG表达主要局限于淋巴细胞亚群(以耗竭CD8+T细胞为著),而TNF则在巨噬细胞和单核细胞中富集(图4d),与B16肿瘤中的发现一致。值得注意的是,Tbk1缺失与对照B16肿瘤的免疫细胞、间质细胞及肿瘤细胞中Tnf和Ifng表达水平相当。这些结果证实ICB后TNF/IFNγ通路被激活,且治疗无应答患者存在持续的细胞因子分泌。
鉴于Tbk1缺失B16肿瘤中免疫区室重塑有限且效应细胞因子表达相当,研究者推测TBK1缺失使肿瘤细胞对TNF/IFNγ敏感性增加。全基因组CRISPR筛选显示,在TNF/IFNγ联合刺激下,Tbk1靶向sgRNA属于最显著耗竭的基因之一(图4e),与B16黑色素瘤体内筛选结果6(图1a)相互印证。体外必需性分析证实Tbk1非必需基因,与其在体外(图1b)和体内(图1c)的生长特性一致。Tbk1缺失B16细胞对TNF/IFNγ联合处理表现显著敏感性,但对单一细胞因子无反应(图4f)。通过单细胞克隆验证发现,该敏感性取决于Tbk1缺失程度:克隆3和4(TBK1完全缺失)细胞活力显著降低,克隆2(TBK1正常表达)无反应,而克隆1(TBK1表达部分保留)呈现部分应答。标准化生长抑制率(GR)分析显示,对照B16细胞仅出现部分生长抑制,而Tbk1缺失细胞在TNF/IFNγ超过阈值浓度时则呈现显著细胞毒效应(图4g)。
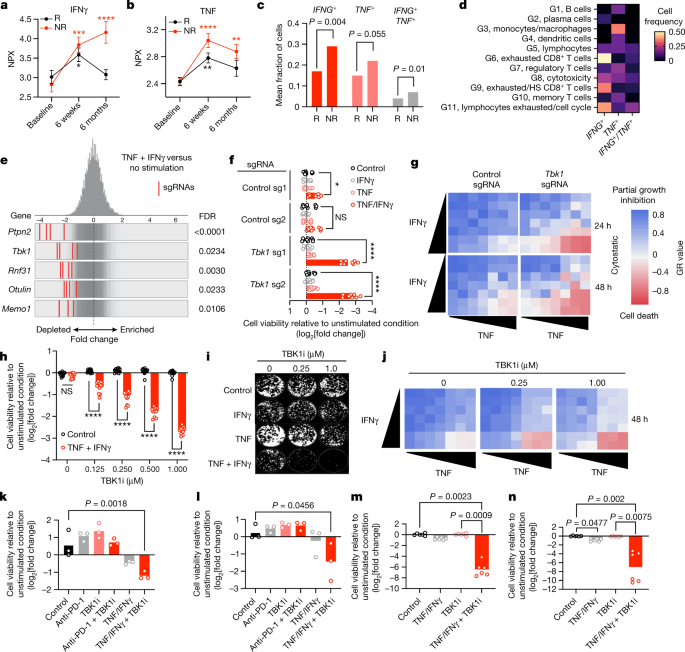
图4:TBK1缺失使肿瘤细胞对TNF/IFNγ敏感化
(6)TBK1抑制剂促进TNF/IFNγ诱导的细胞毒性作用
为探究TBK1药理抑制对细胞活力的影响,,将亲本 B16 细胞用不同浓度的 TBK1i 单独或与 TNF/IFNγ联合处理。结果显示,单用TBK1i(最高1.0μM)对细胞活力无显著影响,而与TNF/IFNγ联用时则呈现剂量依赖性的细胞活力下降(图4h)。TBK1i能显著抑制TNF/IFNγ存在条件下的B16细胞集落形成,对单用TNF的抑制作用较弱(图4i)。该效应在B16-ova细胞及3D培养的亲代B16肿瘤球模型中均得到验证。使用另外两种TBK1抑制剂(MRT6730732和GSK861233)及TBK1靶向蛋白降解嵌合体(TBK1 PROTAC 3i)也观察到类似现象。
生长抑制率(GR)分析证实,在能诱导Tbk1缺失B16细胞产生细胞毒效应的TNF/IFNγ浓度下,TBK1i对亲代B16黑色素瘤细胞具有剂量依赖性作用(图4j)。剂量反应研究表明,单用TBK1i(最高1.0 μM)或分别联合IFNγ/TNF单药,对野生型或Tbk1缺失B16细胞的生长均无影响;而TBK1i与TNF/IFNγ联用时可重现Tbk1缺失细胞对TNF/IFNγ的敏感性。进一步GR分析显示,TNF/IFNγ处理显著增强TBK1i的半数效应浓度(GEC50)及总体药效(GRAOC)。
为在其他模型系统中验证该结果,研究者评估了人源黑色素瘤细胞系、PDOTS及患者来源类器官(PDOs)对TNF/IFNγ的敏感性。与B16细胞类似,TBK1i以时间和剂量依赖方式增敏A375人黑色素瘤细胞对TNF/IFNγ的反应。值得注意的是,获得性BRAF/MEK抑制剂耐药的A375细胞较亲代细胞对TBK1i更敏感。在PDOTS模型(包括PD-1阻断耐药的皮肤型及眼型黑色素瘤)中,TBK1i能显著增强ICB不敏感肿瘤对外源性TNF/IFNγ的应答(图4k,l)。特别值得注意的是,对TBK1i与抗PD-1联合治疗具有显著离体应答的匹配PDOs模型(PDOTS-04和PDOTS-07;图2e,f),同样表现出对TBK1i+TNF/IFNγ的显著敏感性(图4m,n)。这些结果在人与小鼠黑色素瘤细胞系及PDOTS/PDOs等患者来源模型中一致表明:TBK1i治疗可降低肿瘤细胞对TNF/IFNγ的细胞毒性阈值。
(7)TBK1抑制程序性坏死
TBK1通过磷酸化受体相互作用蛋白激酶1(RIPK1)抑制肿瘤坏死因子受体(TNFR)下游的细胞死亡信号通路。TBK1缺失会减弱这种抑制性磷酸化作用,从而促进RIPK1活化,增强TNFR复合体II的形成,进而导致caspase 8剪切和激活。为探究TBK1缺失对RIPK1活化和caspase剪切的影响,研究者对TNF/IFNγ处理的对照和Tbk1缺失B16细胞进行免疫印迹分析。结果显示,Tbk1缺失细胞在TNF/IFNγ处理3小时内即出现磷酸化RIPK1(p-RIPK1, Ser166/Thr189)水平升高,该现象早于caspase 8、caspase 3和PARP的剪切以及c-FLIP降解(图5a)。通过Nec-1s(RIPK1抑制剂)和/或zDEVD-fmk(caspase 3抑制剂)预处理发现,单独抑制RIPK1或caspase 3仅能部分挽救Tbk1缺失细胞的活力下降,而完全阻止细胞死亡需要两者联用(图5b)。使用泛caspase抑制剂(Q-VD-OPh和zVAD-fmk)及caspase-8选择性抑制剂(z-IETD-fmk)也观察到类似结果。
坏死性凋亡是由RIPK1调控、通过激活下游RIPK3和假激酶MLKL介导的细胞死亡形式。与RIPK1研究结果一致,RIPK3抑制剂(HS-1371)38或MLKL抑制剂(GW806742X)联合caspase抑制可保护Tbk1缺失细胞免于TNF/IFNγ诱导的死亡。在克隆形成实验中,抑制RIPK1、RIPK3或MLKL同样能保护亲代B16细胞免受TBK1i联合TNF/IFNγ的杀伤作用。IFNγ处理可转录上调MLKL表达,该效应在Tbk1缺失细胞中更为显著。除pRIPK1和caspase剪切外,Tbk1缺失细胞在TNF/IFNγ处理后还表现出磷酸化(Ser358)和总MLKL水平升高,该现象可被RIPK1抑制剂处理所逆转。
BH3谱分析显示,将线粒体暴露于促凋亡BH3-only肽段(如BIM BH3和PUMA BH3)后,对照组与Tbk1缺失B16细胞的细胞色素c释放无差异。在TNF/IFNγ处理后,TBK1缺失对凋亡启动的影响有限,主要差异源于Tbk1缺失细胞对TNF/IFNγ的反应性发生了改变。对照组与Tbk1缺失细胞对凋亡诱导剂星形孢菌素的敏感性在2D和3D培养中均无显著差异。综上,TBK1缺失并未显著改变细胞凋亡启动或对细胞毒药物的敏感性,但使黑色素瘤细胞更易发生TNF/IFNγ诱导的、依赖RIPK和caspase的细胞死亡。
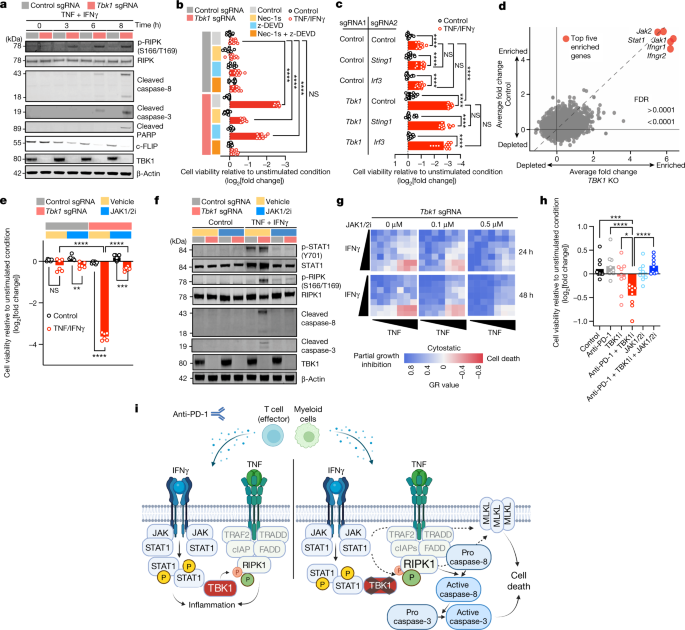
图5:IFNγ信号感知是Tbk1缺失细胞发生RIPK和caspase依赖性死亡的必要条件
(8)STING在程序性坏死过程中是非必需的
TBK1在协调胞浆核酸(如cGAS-STING-TBK1-IRF3-I型干扰素通路)触发的先天免疫应答中具有核心作用,而研究显示对TNF的敏感性增强可驱动cGAS-STING依赖性干扰素反应,进而影响细胞存活。为评估STING-TBK1-IRF3轴的作用,研究者构建了Sting1(亦称Tmem173)和Irf3单独或联合Tbk1缺失的B16细胞系。结果显示,Tmem173与Irf3双敲除B16细胞并未表现出对TNF/IFNγ联合刺激的敏感性增强,而Tmem173或Irf3与Tbk1共同缺失也未改变细胞对TNF/IFNγ的反应性(图5c)。此外,与TNF/IFNγ(无论是否联用TBK1i)不同,STING激动剂(ADU-S100)处理黑色素瘤PDOTS后未显著影响细胞活力。通过多重细胞因子因子检测分析证实,ADU-S100处理可上调CXCL10等炎症因子。结合TBK1缺失细胞中异常的RIPK1活化的发现,这些结果表明Tbk1缺失细胞的TNF/IFNγ依赖性死亡不依赖于胞质核酸感知通路。
(9)完整的IFNγ信号感知能力是必需的
为揭示调控Tbk1缺失细胞对TNF/IFNγ敏感性的关键基因/通路,研究者采用全基因组CRISPR筛选技术,对对照sgRNA和Tbk1 sgRNA B16细胞进行了平行CRISPR筛选。结果显示,靶向IFNγ信号感知相关基因(Ifngr1、Ifgnr2、Jak1、Jak2和Stat1)的sgRNA在两类细胞中均显著富集(图5d),与既往体内外CRISPR筛选结果一致。相比之下,靶向TNFR和坏死性凋亡通路关键组分(如Ripk1、Ripk3、Birc2、Birc3和Casp8)的sgRNA在两组细胞中均未发生显著富集或耗竭,这可能反映了TNFR1下游死亡信号通路调控的复杂性。值得注意的是,对照与Tbk1缺失B16细胞在IFNγ感知通路(JAK1-JAK2-STAT1)的活化、NF-κB(p65)的活化或IRF3的活化方面均无显著差异。
鲁索替尼(JAK1/JAK2抑制剂)预处理可完全保护Tbk1缺失细胞及TBK1i处理的亲代B16细胞免于TNF/IFNγ介导的死亡(图5e)。鲁索替尼处理不仅完全阻断两类细胞的STAT1磷酸化(Tyr701),还消除了Tbk1缺失细胞中RIPK1磷酸化(Ser166/Thr169)、caspase 8和caspase 3剪切(图5f)。生长速率抑制(GR)分析证实,鲁索替尼将Tbk1缺失细胞的细胞毒反应转化为细胞静滞效应,使其反应特征与亲代B16细胞及对照sgRNA B16细胞趋同(图5g)。此外,JAK1/JAK2抑制还能挽救抗PD-1联合TBK1i处理的黑色素瘤PDOTS(图5h)。这些数据证实,Tbk1缺失B16细胞对TNF/IFNγ的增敏效应依赖于完整的IFNγ信号感知,并揭示了IFNγ诱导的JAK-STAT信号通路与TNF介导的RIPK1活化之间存在分子关联(图5i)。
讨论:
该研究揭示TBK1作为免疫逃逸基因,其靶向抑制可通过降低肿瘤细胞对效应细胞因子诱导死亡的敏感性,从而增强PD-1阻断疗效。通过同源小鼠肿瘤模型和患者来源离体模型,研究者证实靶向TBK1能增敏肿瘤对免疫攻击的敏感性。与近期报道的其他免疫逃逸基因不同,肿瘤特异性TBK1缺失并未引起免疫微环境的显著重塑,而是通过增强肿瘤细胞对免疫细胞源性效应细胞因子(TNF与IFNγ)的敏感性发挥作用——这一机制在全基因组CRISPR筛选及后续验证实验中均得到确认。既往研究显示,在缺乏IFNγ信号传导的免疫治疗耐药黑色素瘤细胞中TNF信号发挥关键作用,而研究者的发现则揭示了TNF与IFNγ信号通路间的精密互作,为利用这种互作增敏肿瘤细胞对免疫攻击的应答提供了治疗策略。
尽管多项功能缺失性CRISPR筛选(体内外)均鉴定出TBK1作为潜在免疫逃逸基因,但TBK1抑制能增强癌症免疫治疗应答这一发现仍令人意外。完整的TBK1信号通路是STING激动剂发挥抗肿瘤疗效的必要条件,这类模拟胞质DNA反应的先天免疫刺激分子已被证明可单独或联合免疫疗法抑制肿瘤生长。然而,TBK1在死亡受体信号调控中的这一新兴作用,与其在先天免疫应答和病毒感知中的传统角色截然不同。该研究证明TBK1缺失会导致TNF/IFNγ刺激后发生RIPK和caspase依赖性细胞死亡,并确认STING和IRF3在此肿瘤内在死亡表型中非必需。
虽然TBK1信号缺失不影响免疫缺陷小鼠或分离肿瘤细胞系的生长,但在含自体免疫细胞的模型中,TBK1药理抑制显示出中等抗肿瘤活性,提示其存在肿瘤外在作用机制。与此一致,TBK1i处理可增加肿瘤微环境中早期耗竭/效应CD8+T细胞和M1型巨噬细胞比例,并增强分离CD8+T细胞和巨噬细胞的效应细胞因子表达。因此,TBK1i不仅降低肿瘤细胞对TNF/IFNγ的细胞毒阈值,还促进肿瘤浸润免疫细胞产生这些细胞因子。尽管TNF/IFNγ的系统性上调可能造成组织损伤,但遗传性TBK1缺陷患者仅表现为轻度TNF驱动的自身炎症综合征,而不增加感染风险或严重程度。重要的是,小鼠TBK1i治疗(无论是否联合抗PD-1)均未导致体重下降等系统毒性迹象。未来研究需进一步解析TBK1在不同免疫细胞亚群中的作用,并评估其在免疫治疗耐药黑色素瘤患者中的治疗潜力。
癌症免疫治疗领域面临两大核心挑战:(1)需要能转化至人类免疫系统的临床前模型;(2)缺乏高效评估联合治疗策略的方法。面对逾千项免疫治疗联合试验的开展,亟需新方法来淘汰无效方案并深入理解新疗法的应答与耐药机制。小鼠模型虽便于体内外操作和迭代实验,但缺乏人类肿瘤的异质性;患者来源模型虽具有(人类肿瘤的)异质性和复杂性,但临床相关性更强,并能利用临床相关生物样本评估不同患者间的治疗反应分布。该研究不仅为TBK1靶向治疗策略的研发提供依据,更建立了结合遗传学和药理学工具、跨模型系统评估免疫逃逸靶点的系统性研究框架。

汇报人:吴婷婷
导师:任建君
审核:毛敏姿、李俊虹、任建君
