



精读分享│【Nature】:肿瘤广谱性的RNA 剪接异常产生可用于治疗的公共新抗原
英文题目:Tumour-wide RNA splicing aberrations generate actionable public neoantigens
中文题目:肿瘤广谱性的RNA 剪接异常产生可用于治疗的公共新抗原
期刊:Nature(IF=50.5)
单位:加州大学旧金山分校神经外科系,美国加利福尼亚州旧金山 帕克癌症免疫治疗研究所
发表时间:2025年3月



摘要
基于T细胞的免疫疗法通过靶向癌症特异性抗原,在癌症治疗中展现出巨大潜力。然而,对于体细胞突变较少且肿瘤内异质性显著的肿瘤,其疗效仍面临显著限制。本研究报道了一类来源于多种癌种RNA剪接异常的、此前未被充分认识的肿瘤广谱性的公共新抗原。研究者鉴定出能够识别并靶向GNAS与RPL22基因异常剪接产物的新抗原的T细胞受体克隆型。在多部位肿瘤活检中,研究者观察到GNAS异常剪接位点在胶质瘤、间皮瘤、前列腺癌及肝癌中具有广泛表达。这些新抗原由肿瘤细胞内源性产生并被有效呈递,能够被特异性的CD8⁺ T细胞识别并诱导肿瘤细胞清除。此外,研究者发现特定癌种中剪接因子表达的失调与新剪接位点的重复性上调密切相关。该研究揭示了基于异常RNA剪接产生的公共新抗原作为免疫治疗新靶点的分子基础,为克服肿瘤内异质性带来的治疗挑战提供了新的策略。
引言
基于细胞的免疫疗法为多种恶性肿瘤患者提供了持久的生存获益,但肿瘤内异质性(ITH)导致许多肿瘤逃避免疫清除。尽管免疫疗法在具有高免疫浸润和高突变负荷的肿瘤中效果显著,但在具有广泛ITH或低突变负荷的癌症中常出现治疗抵抗。目前,针对非同义体细胞突变来源的肿瘤特异性抗原(TSAs)的免疫疗法在低突变负荷肿瘤中靶点有限。为拓展治疗选择,近期研究探索了癌症特异性剪接事件(新连接,NJs)作为TSAs的新来源。NJs广泛存在并能产生激活CD8+ T细胞应答的TSAs,但NJs在肿瘤整体(如不同区域或时间点)中的保守性尚未明确,导致其克隆性特征不清。
为填补这一空白,该研究通过分析不同癌种中NJs的克隆性,鉴定出具有跨肿瘤普适性的"公共"NJ来源TSAs。通过建立系统性分析流程,研究者绘制了不同瘤内区域RNA剪接连接图谱,以表征空间保守的NJs(扩展数据图1)。研究发现这些NJ来源的TSAs可经蛋白酶体加工并呈递于高表达的人类白细胞抗原(HLA)分子上,能够激活T细胞受体(TCR)信号通路并诱导CD8+ T细胞依赖的抗原特异性肿瘤杀伤。这些发现表明,靶向肿瘤广谱性公共NJ来源TSAs有望成为一类新型"现成"癌症免疫疗法。
研究路线图
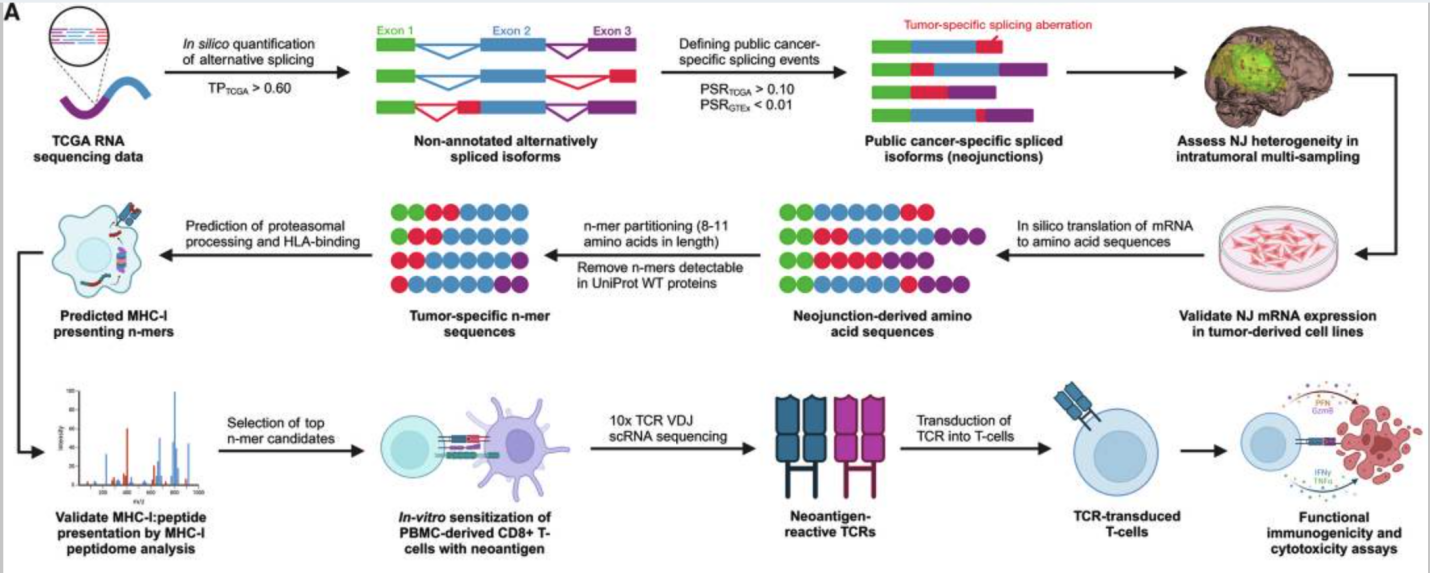
研究主要内容
1. 泛癌NJs的特征
研究者通过对癌症基因组图谱(TCGA)的RNA测序数据进行分析,在12种具有空间定位肿瘤样本的癌症类型中鉴定了未注释的剪接连接点(图1a及扩展数据图1a)。为确保数据可靠性,仅纳入肿瘤纯度≥60%的样本(图1b),并筛选出具有蛋白编码潜力的未注释连接点(扩展数据图1b)。通过定义连接点阳性样本率(PSR,即队列中该连接点表达量≥经典剪接连接点1%的样本占比),研究者在每个TCGA肿瘤队列中筛选出高阳性率的公共新连接点(PSR_TCGA≥10%;图1c及扩展数据图1c)。基于基因型-组织表达(GTEx)计划中9,166例正常组织样本的基线数据(PSR_GTEx<1%;扩展数据图1d),研究者进一步将癌症特异性剪接事件定义为正常组织低表达(PSR<1%)的异常剪接类型。结果显示,平均每个TCGA肿瘤类型可鉴定出94个公共新连接点(图1d及附表1),且其表达频率在不同样本间呈现高度一致性(图1e)。这些公共新连接点具有多样化的剪接类型分布(图1f),且移码诱导型剪接事件的比例保持稳定(图1g)。值得注意的是,部分新连接点与近期剪接研究报道的结果存在重叠(扩展数据图1e,f)。无监督层次聚类分析表明,新连接点的表达模式具有肿瘤类型特异性,提示其可能参与保守的分子机制。此外,跨癌种共表达的新连接点亚群(图1h)的发现,为基于异常剪接事件的泛癌种免疫治疗靶点开发提供了潜在方向。

图1
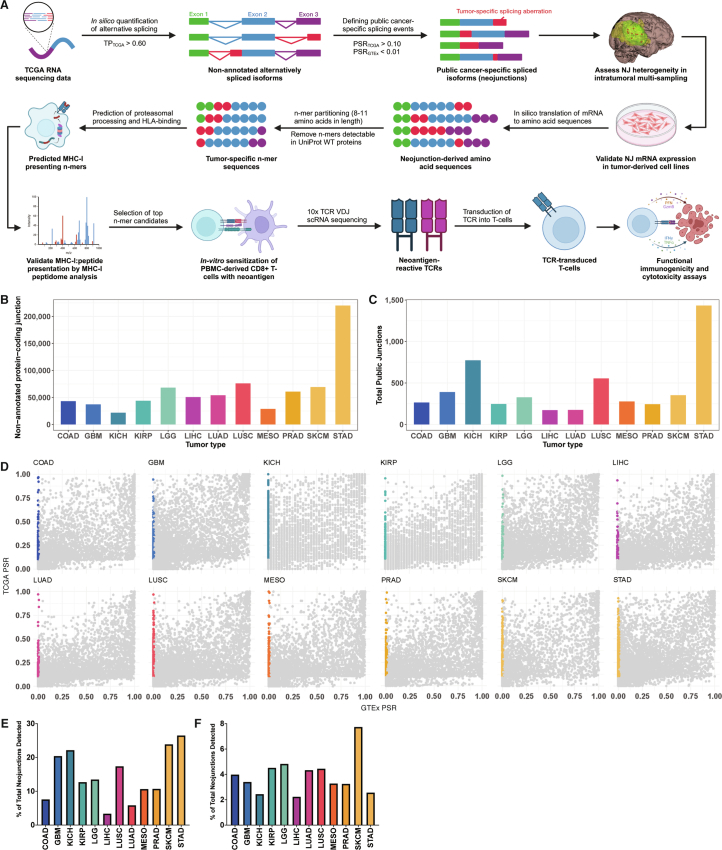
扩展数据图1
2. NJs的肿瘤内异质性
为应对抗原异质性引发的免疫逃逸问题,本研究聚焦于靶向肿瘤全域共有的多新抗原策略(同时靶向多个肿瘤特异性新抗原(neoantigens)的组合策略)。通过对前列腺癌、肝癌、结肠癌、胃癌、肾癌及肺癌等瘤种(图2a,扩展数据图2a)的瘤内RNA测序数据分析,研究者发现公共新连接点(NJs)在多个瘤内区域呈现空间保守性表达(图2b,扩展数据图2b,c),且在大量病例中广泛存在(图2c)。针对胶质瘤这一具有显著瘤内异质性(ITH)特征的肿瘤类型,研究者通过增加样本采样密度(51例胶质瘤病例,每例采集约10个空间分布最大间距的样本进行外显子组及RNA测序分析)系统评估NJs的广谱表达特征(图2d,扩展数据图2d-h)。结果显示,随着样本量从1增加至10,广谱表达NJs的数量与样本量呈显著负相关(扩展数据图2f-h),提示需通过多区域采样策略精准界定肿瘤全域性NJs。
基于大样本瘤内数据分析的无监督层次聚类表明,NJs亚群与异柠檬酸脱氢酶突变型(IDHmut)或野生型(IDHwt)胶质瘤亚型存在特异性关联(图2e)。值得注意的是,IDHmut胶质瘤中肿瘤全域性NJs的数量显著高于IDHwt亚型(图2f),其中45例(88.2%)患者至少检测到1个肿瘤全域性NJs(图2g),13例(25.5%)患者携带超过50个该类NJs(扩展数据图2d)。尽管TCGA数据库注释的774个(98.1%)低级别胶质瘤(LGG)与胶质母细胞瘤(GBM)相关NJs在本研究队列中超过1个瘤区可检测到,但仅37个(4.7%)NJs在超过10%病例的所有样本中持续存在(扩展数据图2e),说明通过多NJs联合靶向可能实现肿瘤全景覆盖。
进一步分析揭示NJs在转移与复发过程中的时空保守性:皮肤黑色素瘤(SKCM)转移灶RNA测序数据显示13个(9.6%)NJs在至少1例患者中跨转移灶持续表达(扩展数据图2c)。TCGA原发-转移配对样本分析显示,结直肠腺癌、前列腺腺癌及SKCM中43.8%-72.6%的原发瘤NJs在转移灶中保留(扩展数据图2i)。类似地,在结直肠腺癌、GBM、LGG、肝细胞癌(LIHC)及肺腺癌(LUAD)的原发-复发配对样本中,平均36.4%的NJs在复发时保持稳定(扩展数据图2j)。胶质瘤队列中,替莫唑胺治疗后复发样本显示79.2%(超突变型)与82.3%(非超突变型)的NJs具有时间保守性(扩展数据图2k),充分验证NJs在肿瘤演进过程中的时空持续性特征。
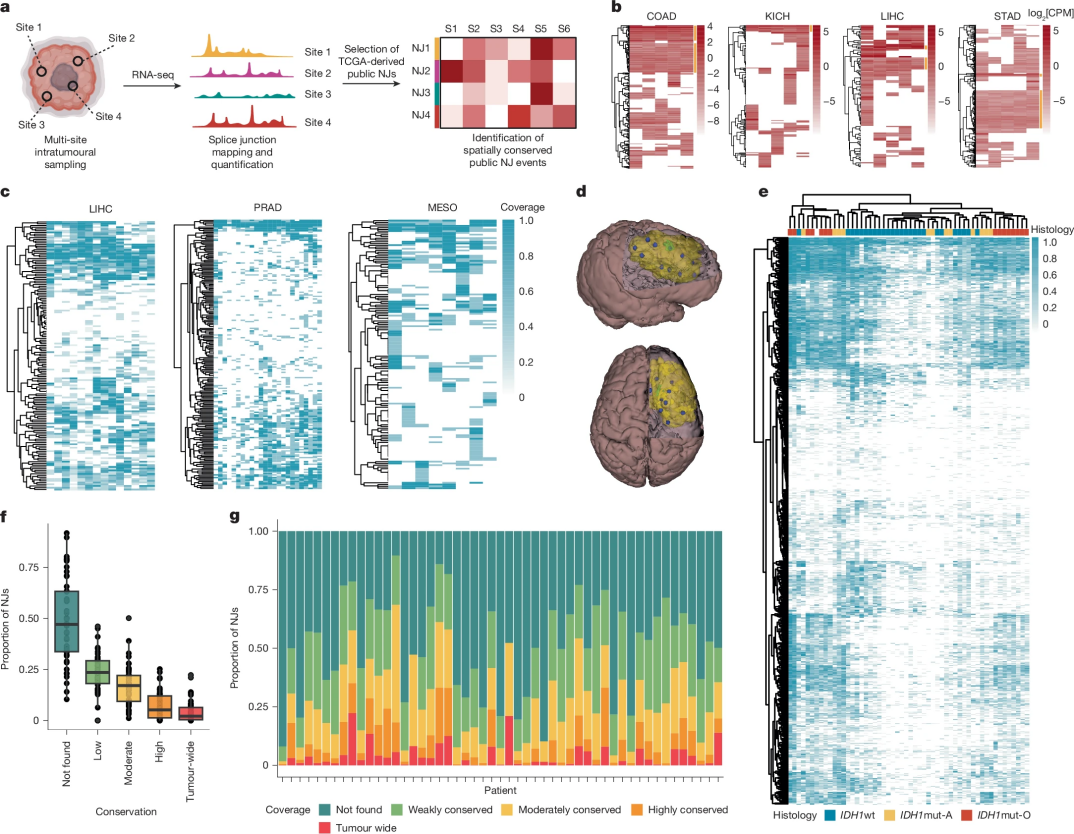
图2
扩展数据图2
3. 肿瘤亚型因素驱动NJ表达
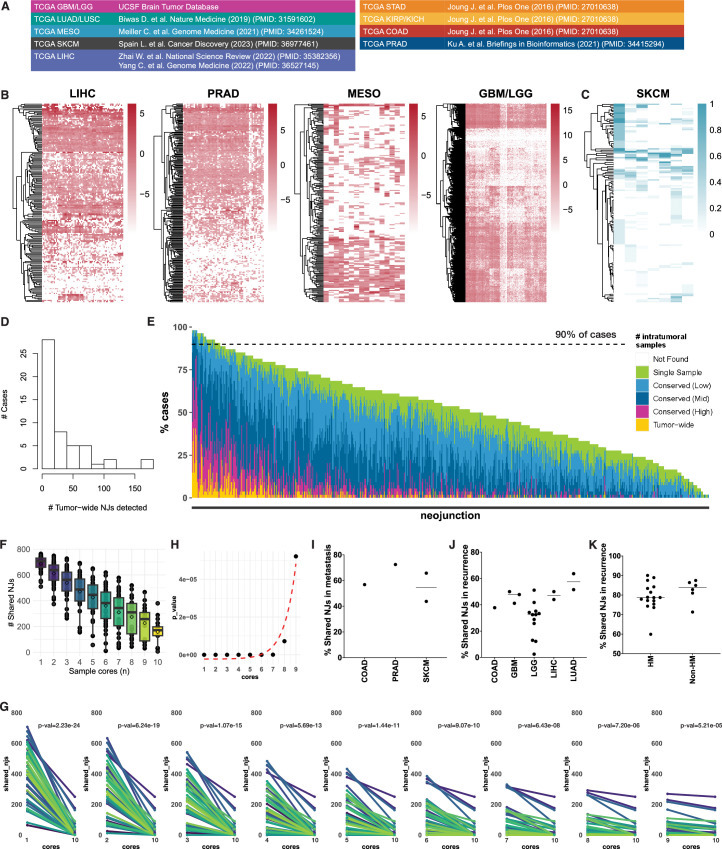
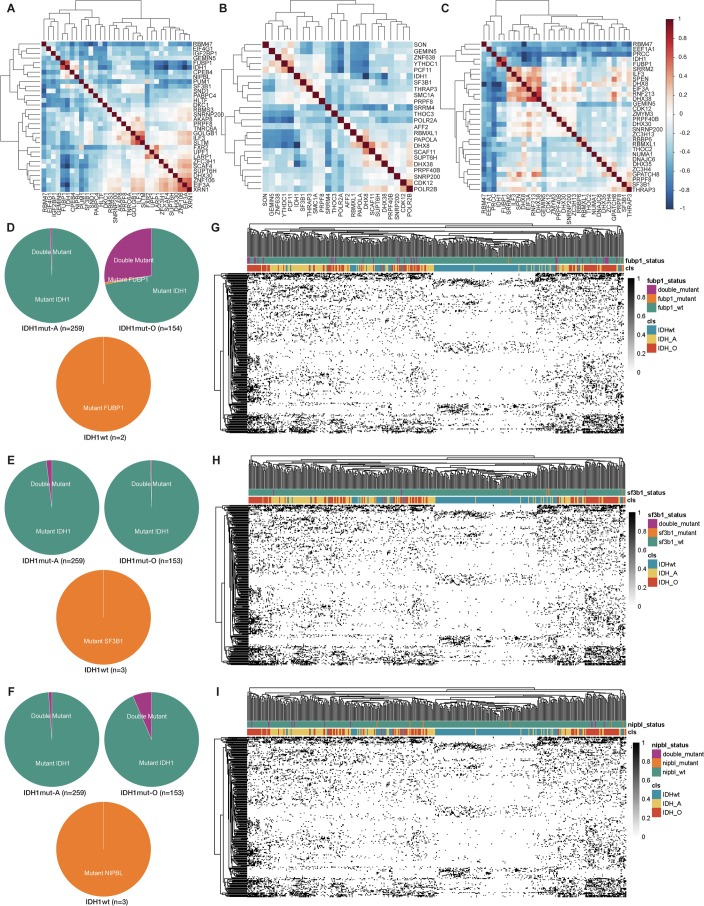
新连接点(NJs)的亚型特异性表达模式(图2e)提示肿瘤亚型相关的剪接机制失调可能参与调控过程。尽管既往研究提示IDH突变可能驱动剪接异常,但研究者发现更复杂的调控网络:在TCGA及本研究的空间多区域测序数据中,IDH突变型(IDHmut)胶质瘤病例的公共NJs数量显著高于IDH野生型(IDHwt)病例(图3a,b)。进一步分析发现,IDHmut亚型中少突胶质细胞瘤(IDHmut-O)的NJs丰度高于星形细胞瘤(IDHmut-A)(图3c,d)。通过成对Pearson相关性分析评估NJs表达与常见RNA剪接因子突变(如FUBP1、SF3A1、NIPBL)的关联性(扩展数据图3a-c),发现FUBP1、SF3A1及NIPBL突变与IDH突变高度共现,且FUBP1突变在IDHmut-O胶质瘤中特异性富集。然而,NJs表达与上述剪接因子突变状态未呈现显著聚类关联(扩展数据图3d-i)。
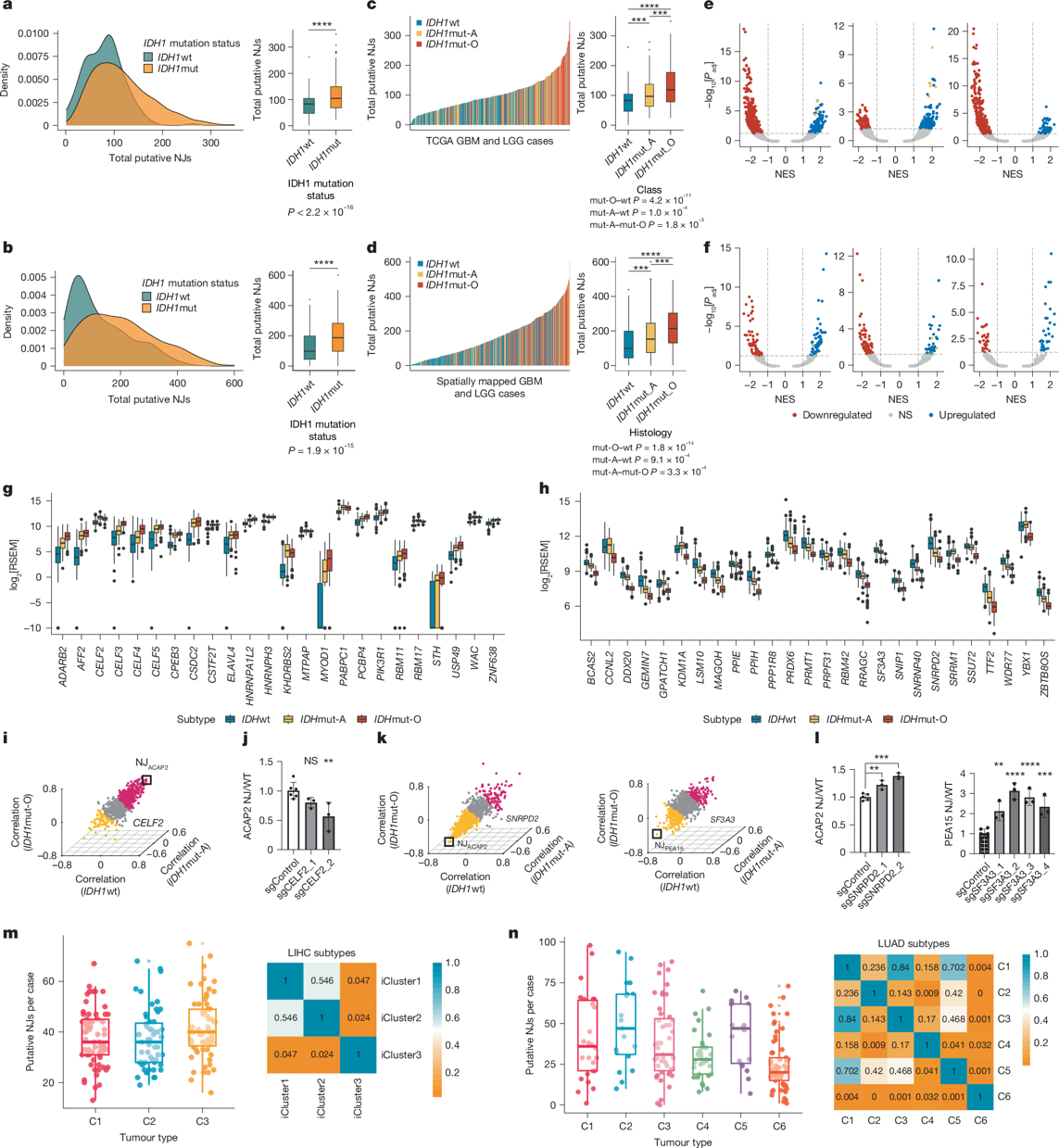
图3
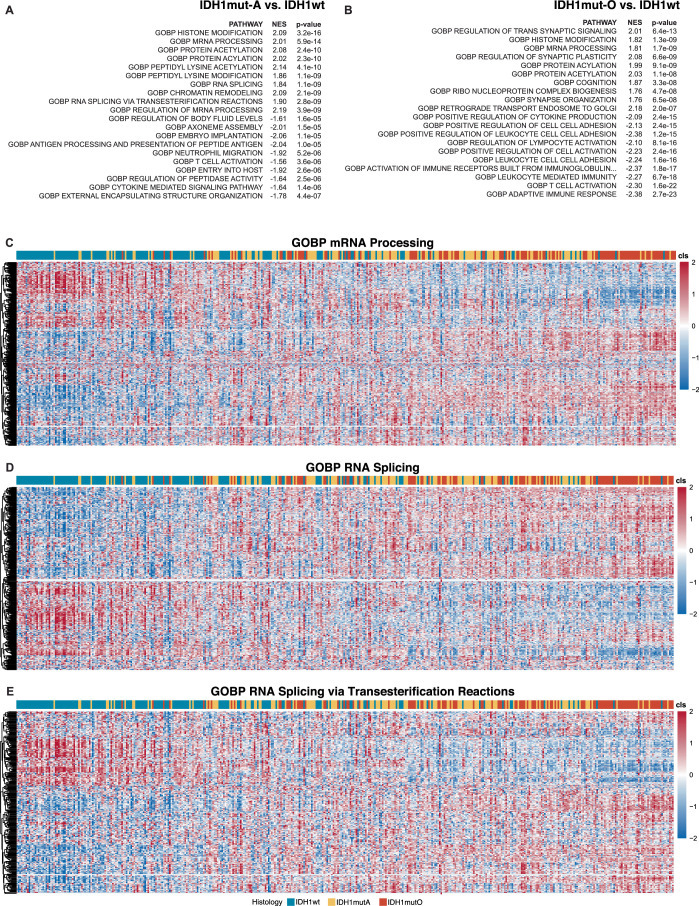
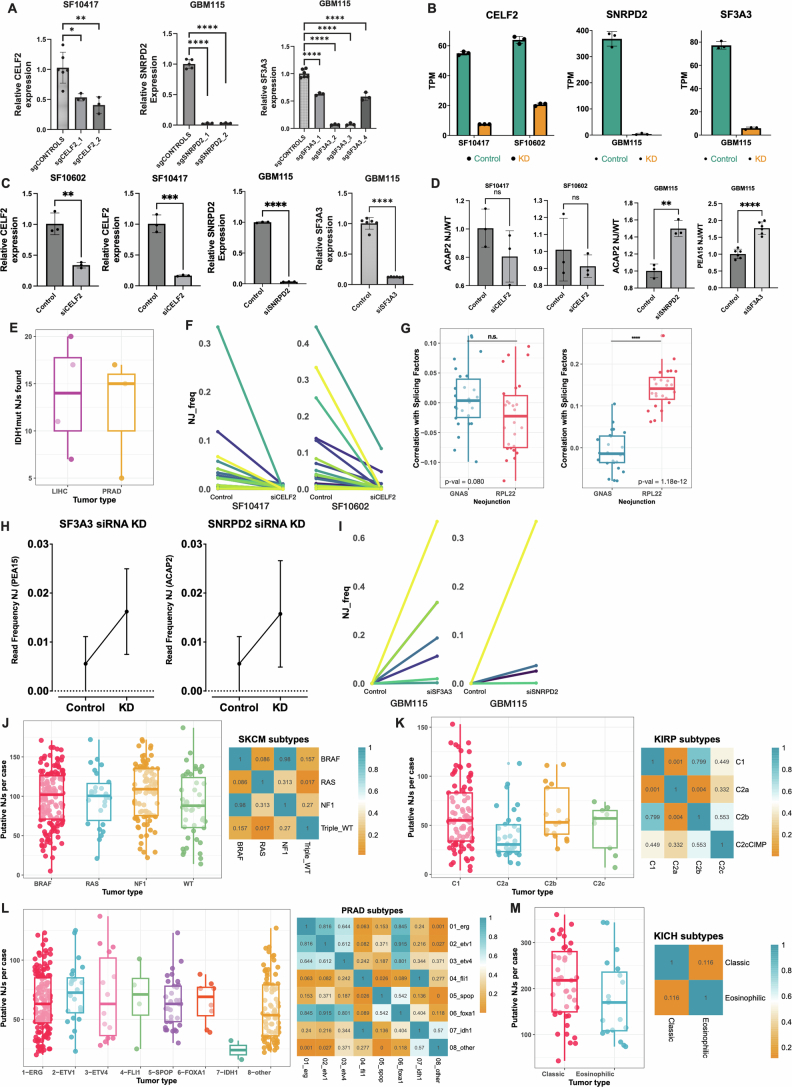
基因集富集分析表明,IDHmut胶质瘤中剪接相关基因集的表达水平在基因本体生物过程(GOBP)及细胞组分(GOCC)数据库中均显著高于IDHwt亚型(图3e,f)。根据NJs表达水平排序,IDHmut亚型中高表达的剪接相关基因呈现显著共表达模块(扩展数据图4c-e),提示其可能驱动亚型特异性NJs生成。筛选GOBP剪接相关基因(n=24)发现,IDHmut病例中CELF2等基因表达量较野生型上调1.5倍以上(P<0.05)(图3g,h)。celf2过表达已被报道可诱导剪接异常,本研究中celf2表达水平与789个公共njs中45.5%(359>0.10),且与NJ-ACAP2相关性最强(图3i)。通过CRISPRi(扩展数据图5a)及siRNA(扩展数据图5b,c)敲低IDHmut细胞系中CELF2表达,发现NJ-ACAP2表达显著降低(图3j;扩展数据图5d)。进一步鉴定出244个在IDHmut胶质瘤中显著上调的NJs(log2[倍数变化]>1.5,P<0.05),其中部分在其他TCGA IDHmut肿瘤中可检测(扩展数据图5e)。CELF2敲低后,少突胶质细胞瘤(SF10417)与星形细胞瘤(SF10602)细胞系中分别有8.6%(19/220)和12.7%(28/220)的IDHmut相关NJs表达下降(扩展数据图5f),且候选NJs表达与IDHmut相关剪接基因表达呈协同变化(扩展数据图5g)。
扩展数据图3
GOBP剪接相关基因集分析(扩展数据图4c-e)显示,IDHmut-O病例中染色体1p/19q共缺失区域内的剪接基因(如SNRPD2、SF3A3)显著下调。筛选26个在IDHmut-O中表达量较IDHmut-A及IDHwt降低1.5倍以上的基因(P<0.05)(图3h),发现SNRPD2与SF3A3的表达水平下降与NJs表达升高显著相关(图3k)。在保留1p/19q双拷贝的GBM115细胞系中敲低SNRPD2或SF3A3,可分别诱导NJACAP2与NJPEA15表达显著升高(图3l;扩展数据图5d,h)。此外,52个IDHmut-O特异性NJs中,7个(13.5%)与4个(7.7%)分别在SF3A3或SNRPD2敲低后表达上调(扩展数据图5i)。上述结果表明,野生型剪接因子表达下调可通过剪接通路异常促进NJs生成。
扩展数据图4
跨癌种分析显示,TCGA肝细胞癌(LIHC)iCluster3与肺腺癌(LUAD)iCluster6亚型呈现显著差异的NJs表达谱(图3m,n)。LUAD iCluster6亚型中23个剪接相关通路持续下调,提示经典剪接基因表达失调可能驱动疾病特异性NJs产生。本研究系统揭示了剪接因子突变以外的全新调控机制,为基于NJs的泛癌种靶向治疗提供了理论依据。
扩展数据图5
4. 公共NEJ衍生RNA与多肽的检测分析
鉴于胶质瘤显著的瘤内异质性(ITH)及其不良预后的特征,本研究进一步在细胞系转录组及肿瘤组织蛋白质组数据中验证公共新连接点(NJs)及其编码蛋白的表达。基于胶质母细胞瘤(GBM)患者来源的异种移植模型(n=66)及低级别胶质瘤(LGG)细胞系(n=2)的RNA测序数据,分别在GBM与LGG中检测到767个(97.2%)及510个(64.6%)公共NJs(扩展数据图6a,b)。为克服批量RNA测序的技术限制,本研究针对部分NJs及其侧翼外显子设计特异性引物,通过深度扩增子测序技术证实胶质瘤细胞系中跨NJs的mRNA转录本表达(扩展数据图6c)。
为验证NJs是否可翻译为功能性蛋白,本研究整合公开的胶质瘤患者(n=447)质谱数据集进行系统性分析,共鉴定到302个(38.3%)公共NJs对应的新肽段(扩展数据图6d)。通过质谱序列特异性检索及谱图验证(扩展数据图6e,f),确认这些肽段跨越异常剪接区域。值得注意的是,41.7%的检测肽段来源于移码突变相关的NJs(扩展数据图6g),提示移码诱导的剪接异常可产生可检测的翻译产物。上述结果表明,NJs相关转录本可有效翻译为蛋白。
综合RNA测序与质谱数据,筛选出192个(24.3%)在全部患者样本中持续表达的公共NJs用于后续研究(扩展数据图6h)。该结果不仅证实公共NJs在胶质瘤中的高度复现性,同时揭示其通过编码肿瘤特异性新抗原参与疾病进程的潜在机制。
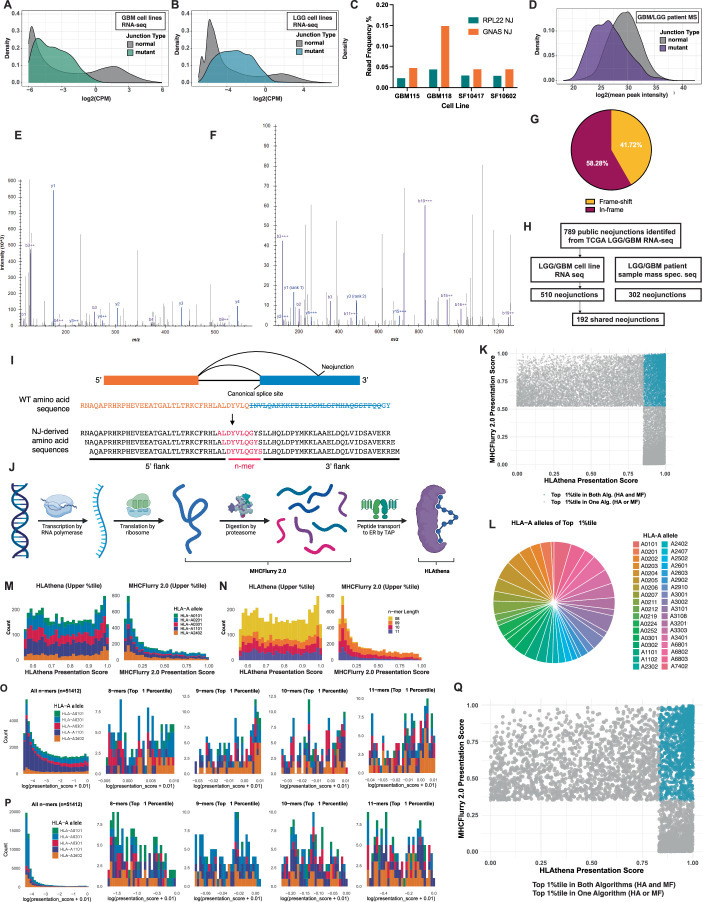
扩展数据图6
5. 肿瘤广泛存在的NJs编码可呈递的新抗原
基于公共新连接点(NJs)可能通过蛋白酶体加工产生可靶向新抗原的假设,本研究系统评估了789个特征性公共NJs生成HLA I类分子递呈肽段的潜力。通过将TCGA数据库中NJs的核苷酸序列进行计算机翻译,构建NJs衍生蛋白数据集,并基于8-11氨基酸长度的多肽片段(扩展数据图6i),筛选出在UniProt正常人组织蛋白质组数据库中未存在的肿瘤特异性多肽。
为预测HLA I类分子递呈的新表位,本研究整合MHCflurry 2.0与HLAthena双算法平台(扩展数据图6j),重点分析了这些多肽与36种高发HLA-A等位基因(扩展数据图6k,l)中HLA-A01:01、HLA-A02:01等五种优势亚型的结合潜能。将两种算法预测结果前1%的高结合多肽(n=832;扩展数据图6m–p)进行交集分析,最终鉴定出315个(39.9%)编码新抗原的NJs(NEJs),其中81个NEJs在转录组与蛋白质组层面均获验证(扩展数据图7g)。值得注意的是,尽管移码突变及外显子3'端异常剪接位点产生的NEJs数量较多(扩展数据图7a,b),其HLA结合评分在不同突变类型间保持相对稳定(扩展数据图7c–f)。
针对北美及欧洲人群高发的HLA-A*02:01等位基因,进一步筛选出32个强结合候选NEJs(扩展数据图7h)。空间多区域测序分析显示,这些NEJs在瘤内呈现高度保守性,尤以GNAS基因座NEJ(NEJ-GNAS,双核苷酸A3缺失型)最为显著(扩展数据图7i)。该发现提示,具有瘤内保守特征的公共NEJs可能通过稳定产生HLA递呈的新肽段,成为泛癌种免疫治疗的潜在靶标。
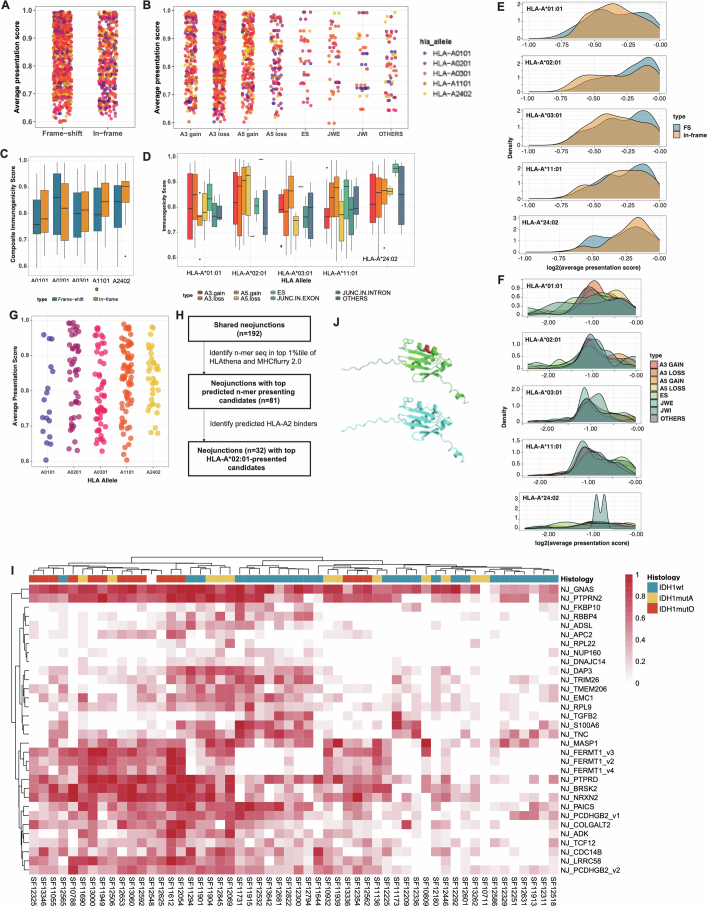
扩展数据图7
6. NEJ反应性TCR的鉴定
为验证NEJ衍生新抗原是否具有驱动T细胞免疫应答的能力,本研究首先通过体外致敏(IVS)技术从健康供体来源的外周血单个核细胞(PBMCs)中筛选新抗原反应性CD8+ T细胞群体(图4a)。实验选取32个NEJ候选新抗原中预测到对HLA-A02:01具有高亲和力结合的4个靶点(扩展数据图6k–m),利用HLA-A02:01阳性健康供体(n=5)的单核细胞来源树突状细胞(moDCs)负载新抗原肽段,与初始CD8+ T细胞共培养以获取特异性TCR基因序列。通过干扰素-γ(IFNγ)酶联免疫吸附试验(ELISA)检测抗原呈递细胞(APC)与CD8+ T细胞共培养体系,发现4个候选新抗原中NeoARPL22和NeoAGNAS可诱导特异性T细胞反应(图4b)。质谱公共数据库验证显示两者均存在对应多肽信号(扩展数据图6e,f)。分子机制分析表明,NeoAGNAS因A3型双核苷酸缺失导致移码突变及提前终止密码子生成,而NeoARPL22为六核苷酸框内缺失,引起α螺旋区域两个氨基酸丢失(扩展数据图7j)。此结果进一步证实人类天然T细胞库中可存在NEJ反应性CD8+ T细胞。
为进一步解析新抗原反应性TCR的分子特征,本研究对NeoARPL22和NeoAGNAS反应性CD8+ T细胞群体进行单细胞V(D)J-转录组联合测序。新抗原特异性TCR克隆型可显著上调IFNG、TNFA及GZMB基因转录水平,且该效应严格依赖于新抗原肽段的存在。实验共鉴定出7个NeoARPL22反应性TCR克隆型(供体3:TCRR3.7、TCRR3.9;供体4:TCRR4.5、TCRR4.6、TCRR4.7、TCRR4.9、TCRR4.11)及1个NeoAGNAS反应性TCR克隆型(供体4:TCRG4.1;图4c)。值得注意的是,TCRG4.1克隆型在CD8+ T细胞群体中扩增至TCR库的4%以上(图4d),表明该新抗原具有强免疫原性。上述发现提示,NEJ衍生新抗原可通过激活特异性TCR克隆扩增驱动抗肿瘤免疫应答,为其临床转化提供关键实验依据。
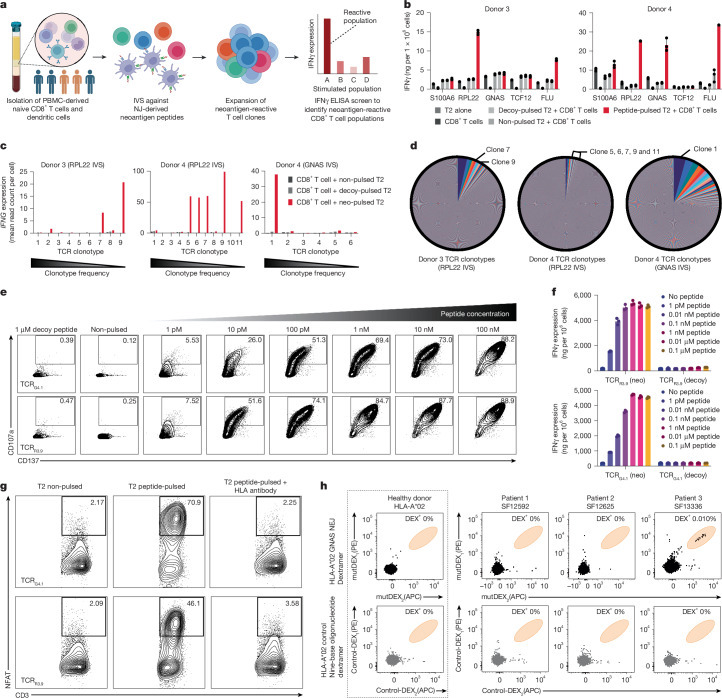
图4
7. NEJ 反应性TCR识别HLA呈递的肿瘤新抗原
为明确TCRR3.9与TCRG4.1克隆型的新抗原特异性反应特征,本研究通过慢病毒载体将上述TCR的α/β链转导至TCR缺陷型三重报告(TR)Jurkat76细胞(表达CD8α–CD8β异源二聚体,即Jurkat76/CD8)或健康供体来源的CD8+ T细胞中(图4e,f)。TR Jurkat76/CD8细胞携带NFAT、NF-κB及AP-1响应元件,可分别驱动eGFP、CFP和mCherry荧光蛋白表达(扩展数据图8a)。与新抗原肽段脉冲化的T2细胞共培养时,TCR转导的TR Jurkat76细胞呈现剂量依赖性激活(扩展数据图8b–d),且两种TCR对新抗原的识别阈值均达到纳摩尔水平,表明其具有较高的功能亲和力。对照肽段(1 μM)的刺激未引发显著TCR活化,进一步验证了其抗原特异性。在PBMC来源的CD8+ T细胞模型中,TCR转导的细胞亦表现出相似的剂量依赖性激活特征。通过检测T细胞活化标志物CD137及脱颗粒标志物CD107a的表达,本研究发现新抗原肽段浓度低至1 pM时即可触发T细胞活化(图4e)。ELISA检测显示,两种TCR的半最大效应肽浓度(EC50)介于0.01–0.1 nM之间(图4f;扩展数据图9a),提示其强效的IFNγ与TNF分泌能力。此外,HLA阻断抗体预处理实验证实,T细胞激活依赖于HLA-A*02:01分子介导的抗原呈递(图4g)。
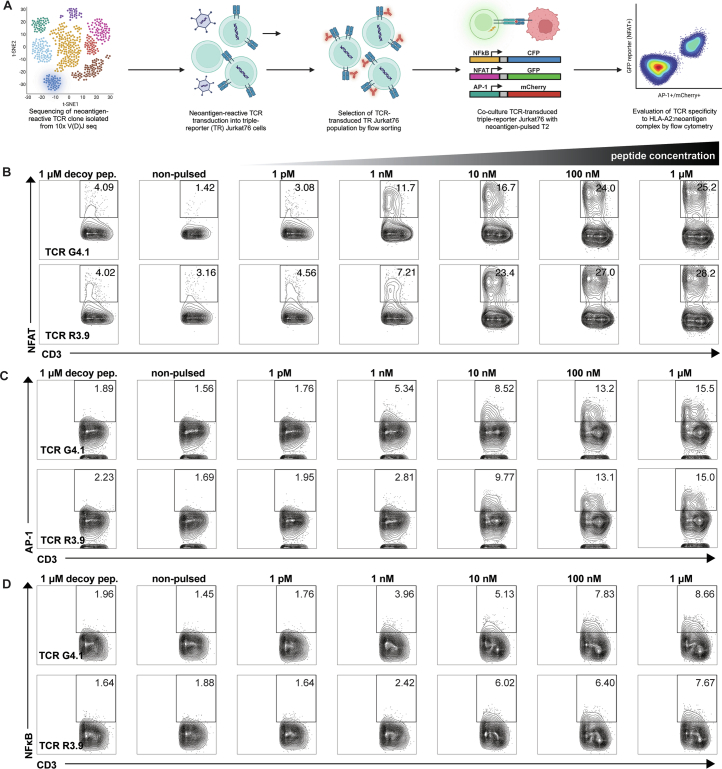
扩展数据图8
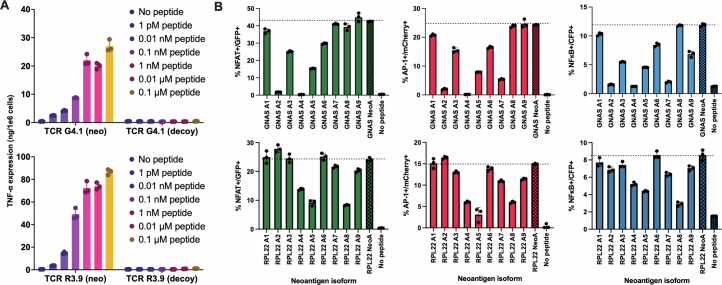
丙氨酸扫描突变分析进一步揭示TCR对新抗原表位的识别关键位点。通过构建残基替换的突变肽段文库,发现TCRR3.9与TCRG4.1对特定氨基酸残基的替换敏感(扩展数据图9b)。基于人类正常蛋白质组数据库(UniProt Proteome ID: UP000005640)的比对分析表明,已知人类蛋白中不存在与TCR识别关键残基相匹配的序列,排除了潜在脱靶风险。上述结果证实,NEJ衍生新抗原特异性TCR具有高灵敏度及精准识别能力,为基于TCR工程化T细胞的免疫治疗策略提供了理论依据。
临床样本验证显示,NEJ反应性CD8+ T细胞可天然存在于表达NEJGNAS的胶质瘤患者(HLA-A02:01阳性)外周血中(扩展数据图6m)。通过短期体外致敏(IVS)实验,3例患者中有1例检测到NEJGNAS特异性T细胞反应,而对照HLA-A02限制性无关新抗原未引发免疫应答(图4h)。该发现不仅支持NEJ衍生新抗原的天然免疫原性,亦为其在肿瘤免疫治疗中的临床应用提供了重要实验证据。
扩展数据图9
8. NEJ 衍生的肿瘤新抗原被加工并呈递给 HLA
为验证NEJ来源转录本是否可生成经HLA功能性呈递并被反应性TCR识别的肽段,本研究通过功能性TCR识别与HLA免疫沉淀联合液相色谱-串联质谱(LC-MS/MS)两种方法评估NEJ衍生新抗原的呈递能力(图5a)。首先,将共转染HLA-A2及全长突变转录本的COS-7细胞与TCR转导的TR Jurkat76或CD8+ T细胞共培养,结果显示TCRR3.9与TCRG4.1转导的TR Jurkat76及CD8+ T细胞均能特异性识别转染对应新抗原的COS-7细胞,证实NEJ来源新抗原可内源性加工并经HLA分子呈递(图5b,c)。随后,对共转染HLA及突变NEJ转录本的COS-7细胞进行HLA-I配体亲和柱免疫纯化,质谱分析鉴定到高丰度的HLA-A2结合肽段NeoAGNAS,且与预测结果一致;同时,在转染HLA-A02:01与NEJRPL22的COS-7细胞中,检测到NeoARPL22两种新抗原肽段,其中九氨基酸多肽的相对丰度更高(图5d)。进一步在未修饰的胶质母细胞瘤细胞系GBM115中检测到HLA-A02:01限制性NeoAGNAS肽段(图5e),表明肿瘤细胞中NEJ的生理表达水平足以生成新抗原。上述实验结果与计算机预测的蛋白酶体加工及HLA结合特性高度吻合(扩展数据图6l),系统性证实了NEJ衍生新抗原的免疫原性及其在肿瘤免疫识别中的关键作用。
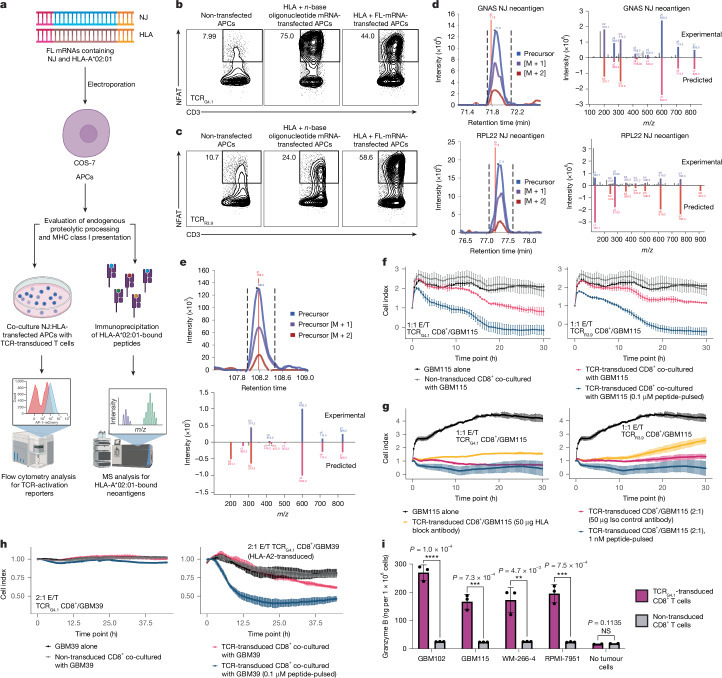
图5
9. NEJ特异性T细胞介导肿瘤细胞毒性
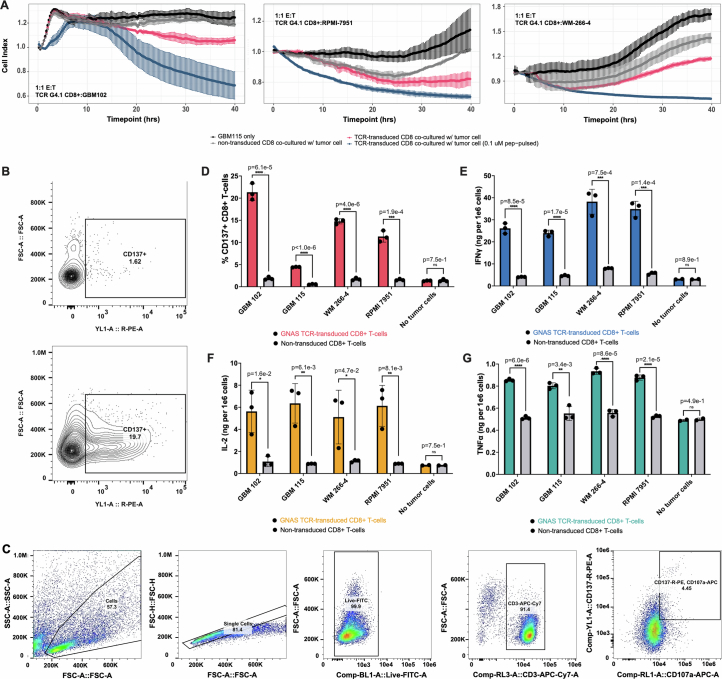
基于已鉴定的新抗原特异性TCR的高敏感性(图4e)及NEJ衍生新抗原的内源性呈递特征(图5e),本研究提出假说:表达公共NEJ的肿瘤细胞可能易受TCR转导T细胞的细胞毒性作用影响。为此,本研究评估了TCR转导的CD8+ T细胞对天然表达NEJRPL22和NEJGNAS的HLA-A02:01阳性肿瘤细胞的杀伤效应。以新抗原肽段脉冲化肿瘤细胞作为阳性对照以定义最大杀伤效率,结果显示,在1:1的效应/靶细胞比例下,TCRR3.9与TCRG4.1转导的CD8+ T细胞可介导TCR依赖性杀伤作用,显著抑制GBM115细胞活性(图5f)。进一步验证中,TCRG4.1转导的T细胞对另一胶质母细胞瘤细胞系(GBM102)及两种黑色素瘤细胞系(RPMI-7951、WM-266-4)展现出同等水平的肿瘤杀伤能力(扩展数据图10a)。HLA-I阻断抗体预处理可部分抑制杀伤效应(同型对照无此作用),证实肿瘤细胞死亡由TCR对HLA-肽复合物的特异性识别所触发(图5g)。在HLA-A2阴性但表达NEJGNAS的胶质母细胞瘤细胞系(GBM39)共培养实验中,TCRG4.1转导的T细胞仅当靶细胞被转染HLA-A02:01基因时才表现出显著细胞毒性(图5h),明确提示NEJ依赖性杀伤需HLA分子介导。与非转导T细胞相比,TCR转导的CD8+ T细胞与肿瘤细胞共培养后表面CD137表达上调(扩展数据图10b–d),进一步支持新抗原特异性T细胞活化。此外,相较于未转导组,TCR转导T细胞分泌的颗粒酶B水平显著升高(图5i),揭示了新抗原特异性细胞毒性的分子机制;而干扰素γ(IFNγ)、白细胞介素-2(IL-2)及肿瘤坏死因子(TNF)分泌量增加(扩展数据图10e–g),则系统性佐证了CD8+ T细胞的新抗原特异性激活。综上,本研究证实NEJ衍生新抗原可被内源性加工并以充足水平呈递,从而通过特异性T细胞介导高效的肿瘤杀伤,为基于TCR工程化治疗的临床转化提供了关键实验证据。
扩展数据图10

汇报人:程丹妮
导师:赵宇
审核:冯兰、任建君
