



精读分享│【Cell Reports】:CHK2-USP7轴的磷酸化-去泛素化正反馈回路在氧化应激下稳定p53
英文题目:The phosphorylation-deubiquitination positive feedback loop of the CHK2-USP7 axis stabilizes p53 under oxidative stress
中文题目:CHK2-USP7轴的磷酸化-去泛素化正反馈回路在氧化应激下稳定p53
期 刊:Cell Reports (IF=7.5)
单 位:中国医科大学曹流教授团队
时 间:2024年6月25日

引言:
p53是一个众所周知的肿瘤抑制蛋白,在DNA损伤和氧化应激条件下,p53调节多种信号通路并维持细胞稳态。泛素特异性蛋白酶7(USP7)调节参与 DNA损伤修复、肿瘤发展和免疫的各种蛋白质的稳定性,是p53通路的关键调控因子。USP7因其在调节p53和抗肿瘤反应中的关键作用而受到广泛关注。然而,在氧化应激下激活USP7-p53轴的潜在机制仍不清楚。
总体研究思路:
主要结果:
1、CHK2与USP7的相互作用
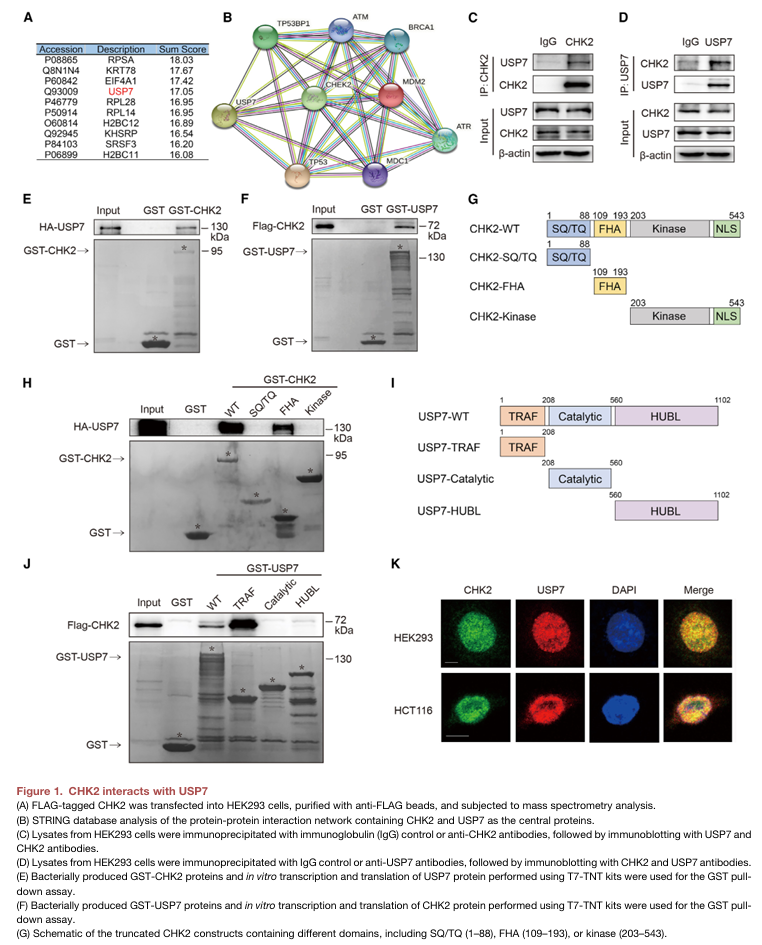
先前已发现USP7通过去泛素化促进p53稳定性。然而,USP7-p53通路的激活机制仍不清楚。作者对纯化的CHK2蛋白进行的质谱分析揭示了其与USP7的潜在相互作用(图 1A)。此外,使用STRING数据库的分析还表明CHK2和USP7之间可能存在蛋白质-蛋白质相互作用(图1B)。作者通过在HEK293(图1C和1D)和HCT116细胞中进行免疫共沉淀(coIP) 分析证实了CHK2和USP7之间的相互作用。体外谷胱甘肽S-转移酶(GST) -pulldown 测定进一步证实了USP7和CHK2之间的直接相互作用(图1E和1F)。为了确定CHK2和USP7 之间相互作用所需的特异性结构域,构建了各种融合蛋白。首先,GST-CHK2 融合蛋白(每个蛋白包含一个特定结构域)用于USP7的GST沉降测定(图1G)。含有FHA结构域的 GST-CHK2 融合蛋白可以与USP7相互作用(图1H),表明FHA结构域在USP7-CHK2 相互作用中的重要作用。同样,在GST pull-down测定中测试了每个包含不同结构域的GST-USP7 融合蛋白(图1I)。包含TRAF结构域的GST-USP7融合蛋白能够与CHK2相互作用(图1J),表明USP7的TRAF 结构域是与CHK2结合的特异性需要。除了体外结合测定外,作者还通过使用共聚焦显微镜的免疫荧光分析观察到USP7和CHK2在HEK293和HCT116细胞中的共定位(图1K)。总之,这些结果证实了CHK2与USP7的相互作用。
2、CHK2磷酸化USP7的S168和T231位点并促进p53去泛素化和稳定性
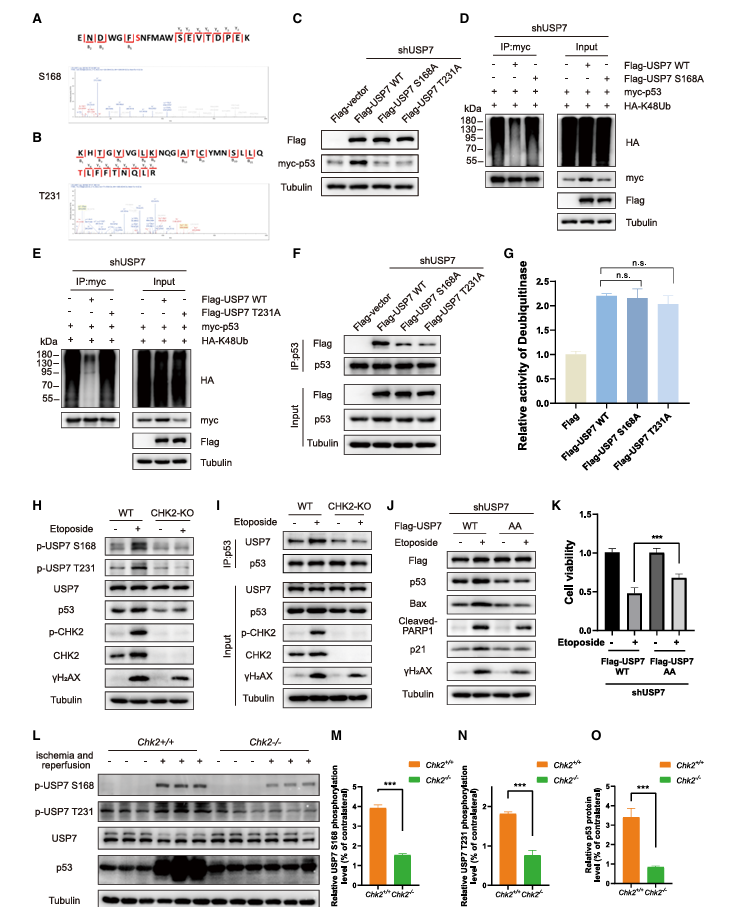
为了确定CHK2是否能磷酸化USP7并激活其去泛素化功能,作者使用纯化的GST标记的USP7蛋白进行了体外CHK2激酶实验,然后对磷酸化位点进行了质谱分析。结果显示,丝氨酸(S)168和苏氨酸(T)231是CHK2激酶的USP7磷酸化位点(图2A-B)。随后,构建USP7 S168和T231A突变体进行突变分析,WB实验和泛素化IP实验表明USP7的S168A和T231A突变体无法稳定p53蛋白,表明USP7的磷酸化对于去泛素化和稳定p53至关重要。此外,USP7(S168A)和USP7(T231A)突变体减少了USP7和p53之间的相互作用(图 2F),而 USP7(S168A)和USP7(T231A)突变体没有显着改变USP7的DUB活性。DNA损伤刺激会增加p53蛋白的水平。用依托泊苷或过氧化氢刺激后,HEK293细胞中细胞内p53 蛋白水平增加。这种增加会被ROS清除剂NAC处理所抑制。USP7的敲低降低了氧化应激下 p53的增加,突出了USP7在氧化应激期间稳定p53中的关键作用。用依托泊苷处理含有 CHK2蛋白稳定敲除(HEK293 CHK2-knockout [KO])的细胞。依托泊苷刺激显着增加了 HEK293 WT细胞中USP7磷酸化和p53的水平,而在HEK293 CHK2-KO细胞中未观察到显著增加(图 2H)。CoIP分析结果显示,依托泊苷刺激后,HEK293 WT细胞中p53与USP7的结合增强,但在HEK293 CHK2-KO细胞中p53和USP7之间的相互作用仍然较弱。当用依托泊苷处理过表达USP7的细胞时,p53、p21、Bax和裂解的PARP1蛋白水平升高。然而,FLAG-USP7-2A(S168A/T231A)突变体的过表达并没有响应依托泊苷刺激而增加p53、p21、Bax和裂解的PARP1的蛋白水平(图 2J)。这表明 USP7 S168和T231位点的磷酸化是观察到的p53响应应激条件增加所必需的。此外,根据CCK-8测定,与依托泊苷处理下的FLAG-USP7 AA(S168A/T231A)突变体相比,过表达FLAG-USP7 WT的细胞存活率降低。
作者还研究了CHK2在USP7磷酸化中的作用及其对p53蛋白稳定性的影响。构建体内小鼠中风模型,因为脑缺血触发了一系列复杂的DNA损伤事件,导致ROS诱导的组织损伤。WB分析显示,p53水平和USP7 S168和T231的磷酸化水平在WT小鼠中显著升高,而在Chk2/小鼠中没有升高。这些数据结果表明CHK2-USP7磷酸化途径有助于p53蛋白的稳定。
3.USP7可以稳定CHK2蛋白的表达
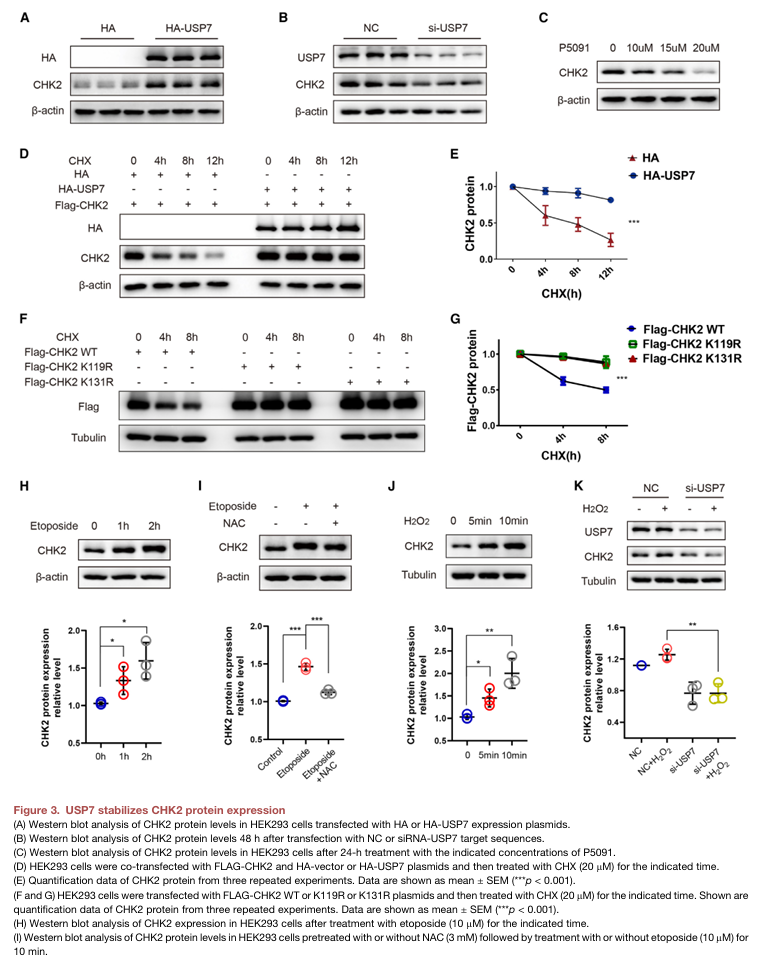
USP7可通过去泛素化防止各种蛋白酶体降解,从而稳定它们的表达。因此,作者推测USP7也可能稳定CHK2蛋白。作者通过过表达USP7并观察到HEK293细胞中内源性CHK2蛋白质水平的升高。敲降USP7会导致HEK293细胞内源性CHK2蛋白的下降,用USP7的特异性抑制剂P5091处理后,两种HEK293细胞均以浓度依赖的方式降低了细胞内CHK2蛋白水平(图3A-C)。
为确定USP7导致CHK2去泛素化的潜在位点,进行了在线数据库搜索,鉴定K119和 K131是CHK2蛋白上的潜在泛素化位点。作者的研究发现CHX处理HEK293细胞,过表达CHK2 K119R和K131R延长了CHK2蛋白的半衰期,提示K119和K131可能是CHK2蛋白的泛素化位点。表明USP7增强了CHK2蛋白的稳定性;此外,研究发现USP7是氧化应激条件下稳定CHK2蛋白的关键蛋白。
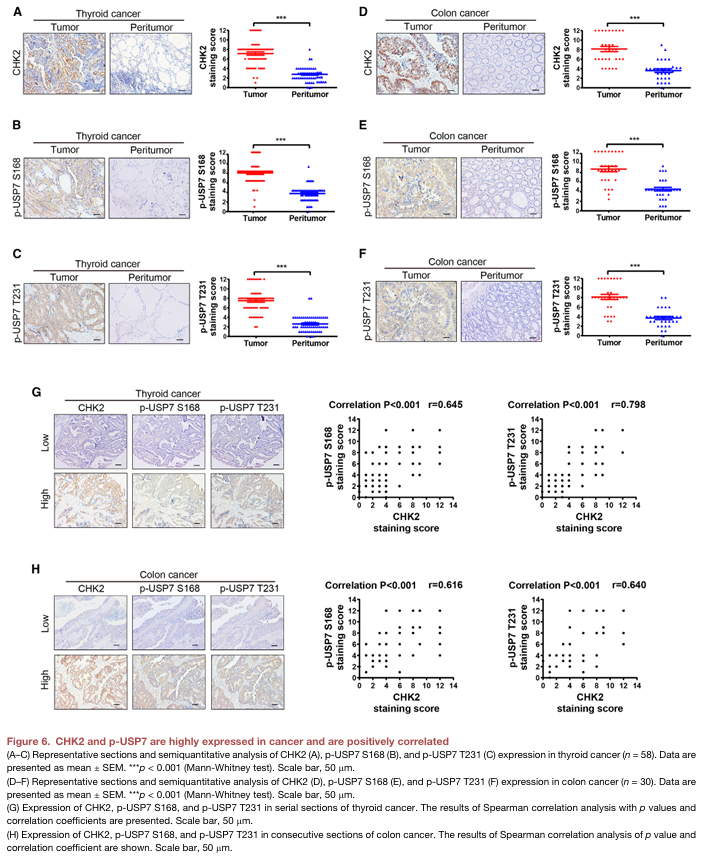
4.USP7去泛素化CHK2 K48连接的K119和K131的多泛素化
为探究USP7蛋白是否通过去除CHK2蛋白泛素化来提高CHK2的蛋白水平,作者在HEK293细胞中过表达FLAG-USP7、myc-CHK2和HA-K48Ub。USP7过表达显著降低了CHK2蛋白的K48连接的多泛素化,敲降USP7显著增加了CHK2的泛素化。用USP7的特异性抑制剂P5091处理HEK293细胞,导致CHK2泛素化显著增加,CHK2蛋白水平显著降低。以上结果表明USP7通过去泛素化来稳定CHK2蛋白(图4A-C)。作者的研究进一步显示当赖氨酸(K)K119或K131被精氨酸(R)取代时,USP7的过表达对CHK2蛋白水平的影响不大(图4D-E)。coIP分析显示,过表达USP7并没有降低CHK2 K119R或K131R突变体的K48连接的泛素化水平(图F、G),而不是对WT CHK2的泛素化水平。这些结果表明,K119和K131是USP7所靶向的CHK2蛋白的特异性去泛素化位点。为进一步验证USP7介导的泛素化是否有助于CHK2蛋白的稳定。作者在HEK293细胞中发现USP7抑制了氧化应激CHK2的增加,并降低CHK2 K48连接的泛素化。综上,这些结果表明USP7在氧化应激条件下通过去泛素化K119和K131位点的CHK2 K48连接的多泛素化来稳定CHK2蛋白水平。
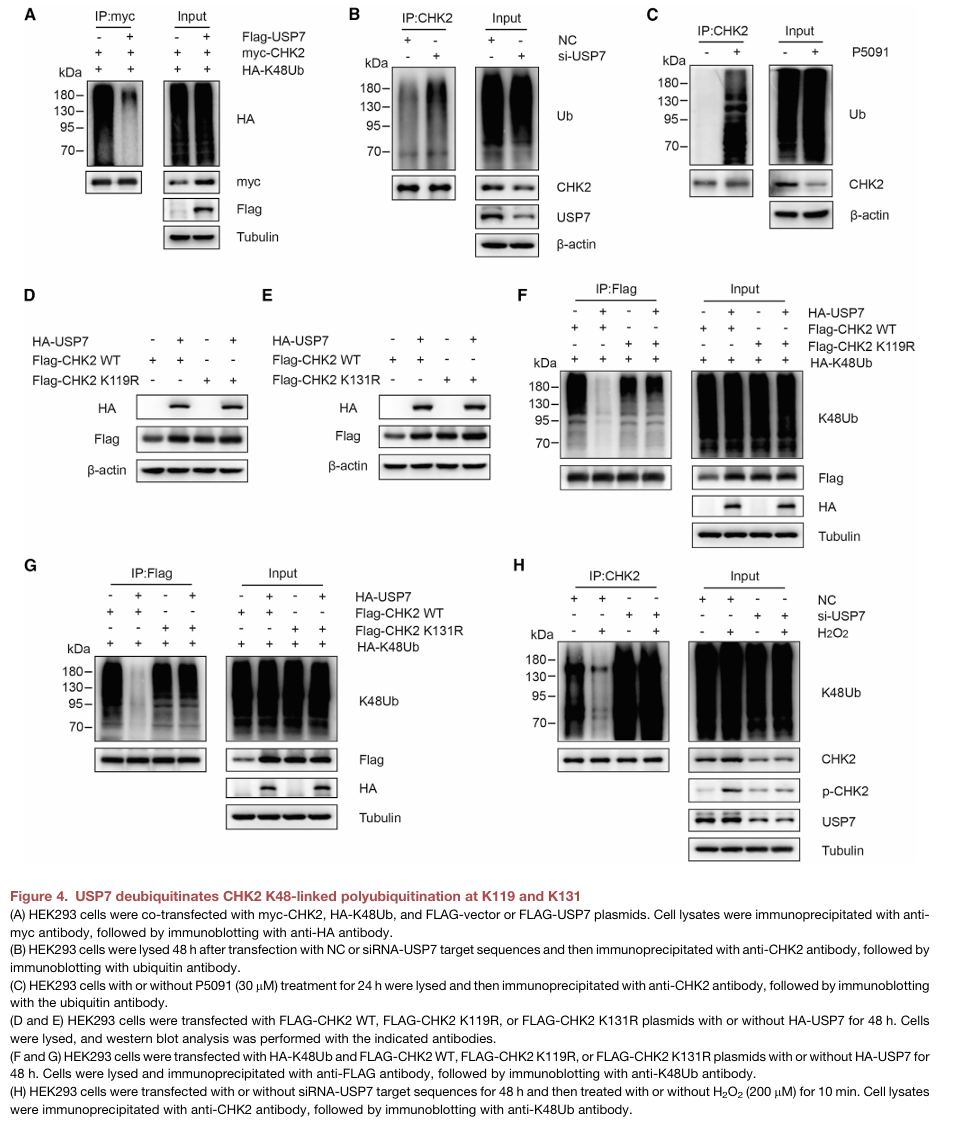
5.CHK2磷酸化USP7也能促进CHK2的稳定,在氧化应激下形成一个正反馈回路
作者探究了CHK2介导的USP7磷酸化是否对USP7增强CHK2蛋白的稳定性的必要性。作者的研究发现将USP7 S168或T231突变为丙氨酸,可消除USP7对CHK2稳定的影响。当USP7 (S168A) 或USP7 T231A突变体在HEK293 shUSP7细胞中过表达时,不能去除CHK2 K48连接的泛素化并提高其稳定性(图5A-C)。
ATM对CHK2 T68的磷酸化对CHK2发挥其功能很重要;因此,使用T68D(模拟CHK2 Thr68磷酸化)和T68A(模拟无Thr68磷酸化)来探讨CHK2 T68磷酸化是否促进其与 USP7的相互作用。发现无论是否处于应激状态,T68D都促进了CHK2与USP7的相互作用(图5D、E)。此外,作者的研究发现内源性USP7的Ser168/Thr231的磷酸化在氧化应激条件下显著增强,但在CHK2-KO HEK293细胞中没有显著变化。(图5F)
USP7(S168A)和USP7(T231A)突变体减少了USP7和CHK2之间的相互作用(图5H)。而依托泊苷促进USP7和CHK2蛋白的相互作用。使用抗氧化剂NAC处理阻断了依托泊苷诱导的p-USP7 Ser168/Thr231的增加,这表明ROS是这一过程的关键信号分子。在HEK293细胞中,ATM抑制剂KU-55933预处理抑制了依托泊苷或过氧化氢诱导的p-USP7 S168、p-USP7 T231和CHK2蛋白水平的升高(图5J-L)。多色免疫荧光染色测定表明 DNA 损伤后pCHK2 T68和p-USP7 S168或T231共定位。
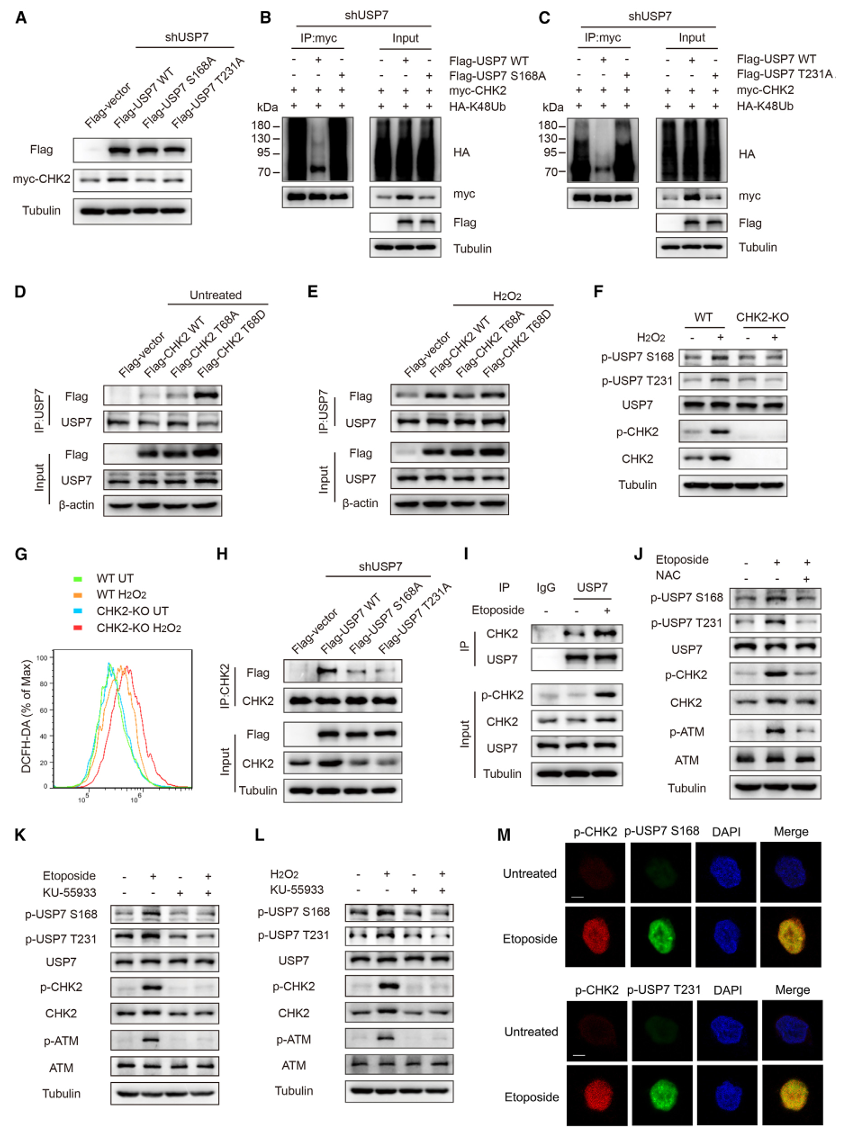
6.CHK2和p-USP7在癌症中高表达,并呈正相关
肿瘤发生过程伴随着ROS的增加,ROS诱导DNA损伤并激活DDR 通路。为了研究 CHK2-USP7轴是否在癌组织中被激活,作者进行了免疫组织化学分析,以评估CHK2和 USP7在S168和T231位点的磷酸化在癌组织及其相应的非癌组织中的表达。据报道,ROS 和DDR与结肠癌和甲状腺癌有关。研究共纳入58例甲状腺癌和30例结肠癌。结果显示,与成对的瘤周组织相比,CHK2、p-USP7 S168和 p-USP7 T231在甲状腺癌和结肠癌组织中均高度表达(p<0.001)(图6A-6F)。重要的是,CHK2的表达水平与甲状腺癌组织中 p-USP7 S168(p<0.001,r=0.645)和p-USP7 T231(p<0.001,r=0.798)的水平呈正相关(图6G)。同样,CHK2的表达水平与结肠癌组织中p-USP7 S168(p<0.001,r=0.616)和 p-USP7 T231(p<0.001,r=0.640)的水平呈正相关(图6H)。这些发现表明,在肿瘤发生过程中,涉及 CHK2-USP7 轴的磷酸化-去泛素化正反馈回路被激活。
结论:
该研究发现ROS-ATM-CHK2在S168/T231位点磷酸化USP7,这对于其在p53上的去泛素化活性至关重要。反过来,USP7在K119/K131位点使CHK2去泛素化,稳定CHK2并形成正反馈回路。这项研究表明,CHK2和USP7之间存在磷酸化-去泛素化正反馈回路,导致p53的稳定和细胞稳态的维持。研究的局限性在于虽然作者发现USP7去除了CHK2在K119 和K131位点的K48连接的多泛素化,但尚不清楚CHK2是否有其他泛素化位点。此外,p-USP7与癌症患者预后的关系尚不清楚。p-USP7是否可以成为与癌症患者的临床参数和预后相关的生物标志物也有待后续研究。

汇报人:郝智贞
导师:刘锋、刘吉峰
审核:肖瑶、任建君
