


原创 邹宇豪 华西医院耳鼻喉科
华西耳鼻喉学术前沿速递——文献精读(第62期)
精读分享│【Cancer Discovery】:胰腺癌对致癌 KRAS 抑制剂产生耐药性的机制
英文题目:Mechanisms of Resistance to Oncogenic KRAS Inhibition in Pancreatic Cancer
中文题目:胰腺癌对致癌 KRAS 抑制剂产生耐药性的机制
期刊:Cancer Discovery(IF: 29.7)
单位:丹娜-法伯癌症研究所肿瘤内科,波士顿,马萨诸塞州
发表时间:2024年11月1日



摘要:
KRAS抑制剂在胰腺导管腺癌(PDAC)中展现出临床疗效,但其耐药性较为常见。本研究发现,在接受阿达格拉西布或索托拉西布治疗的KRASG12C突变PDAC患者中,耐药者出现了PIK3CA和KRAS突变,以及KRASG12C、MYC、MET、EGFR和CDK6基因的扩增。此外,在接受KRASG12D抑制剂MRTX1133治疗的PDAC细胞系和类器官模型中,上皮-间充质转化(EMT)和PI3K-AKT-mTOR信号通路的激活被发现与耐药相关。在KrasLSL-G12D/+; Trp53LSL-R172H/+; p48-Cre(KPC)小鼠模型中,MRTX1133治疗引起了肿瘤的显著缩小。但是,MRTX1133治疗最终出现了耐药,并伴随着Kras、Yap1、Myc、Cdk6和Abcb1a/b的基因扩增以及耐药相关转录程序的共同进化。此外,在KPC和患者来源异种移植(PDX)模型中,间充质样和基底样细胞状态相比经典细胞状态对KRAS抑制剂更敏感。最后,在PDAC小鼠模型中,KRASG12D抑制剂与化疗联合治疗显著改善了肿瘤控制率。这些数据揭示了KRAS抑制剂耐药过程中共同演化的耐药机制,并支持多种联合治疗策略的开发。
研究重要性:
获得性耐药可能会限制KRAS抑制剂在胰腺导管腺癌(PDAC)患者中的疗效。通过临床样本和多种临床前模型,本文揭示了KRAS抑制剂耐药的异质性遗传和非遗传机制,这些发现可为联合治疗策略提供指导,从而提高这些有前景疗法的疗效和持久性,造福患者。
研究背景:
胰腺导管腺癌(PDAC)是一种危重疾病,大多数患者确诊时已是晚期,预后极差,超过90%的患者在诊断后12个月内死亡。KRAS突变在90%以上的患者中存在,是PDAC的重要治疗靶点,但现有KRAS抑制剂疗效有限且耐药普遍出现。KRASG12D抑制剂(如MRTX1133)在临床前研究中展现出显著疗效,亟需深入解析其在PDAC中的治疗反应与耐药机制,为优化联合治疗策略提供依据。
研究目的:
本研究旨在揭示KRAS抑制剂在胰腺导管腺癌(PDAC)中的治疗反应与耐药机制,解析其遗传和非遗传耐药特征。通过临床样本与多种细胞动物模型,探索耐药的潜在机制,为开发更有效的联合治疗策略提供理论基础和实验依据,以改善PDAC患者的治疗效果和疗效持续性。
主要结果:
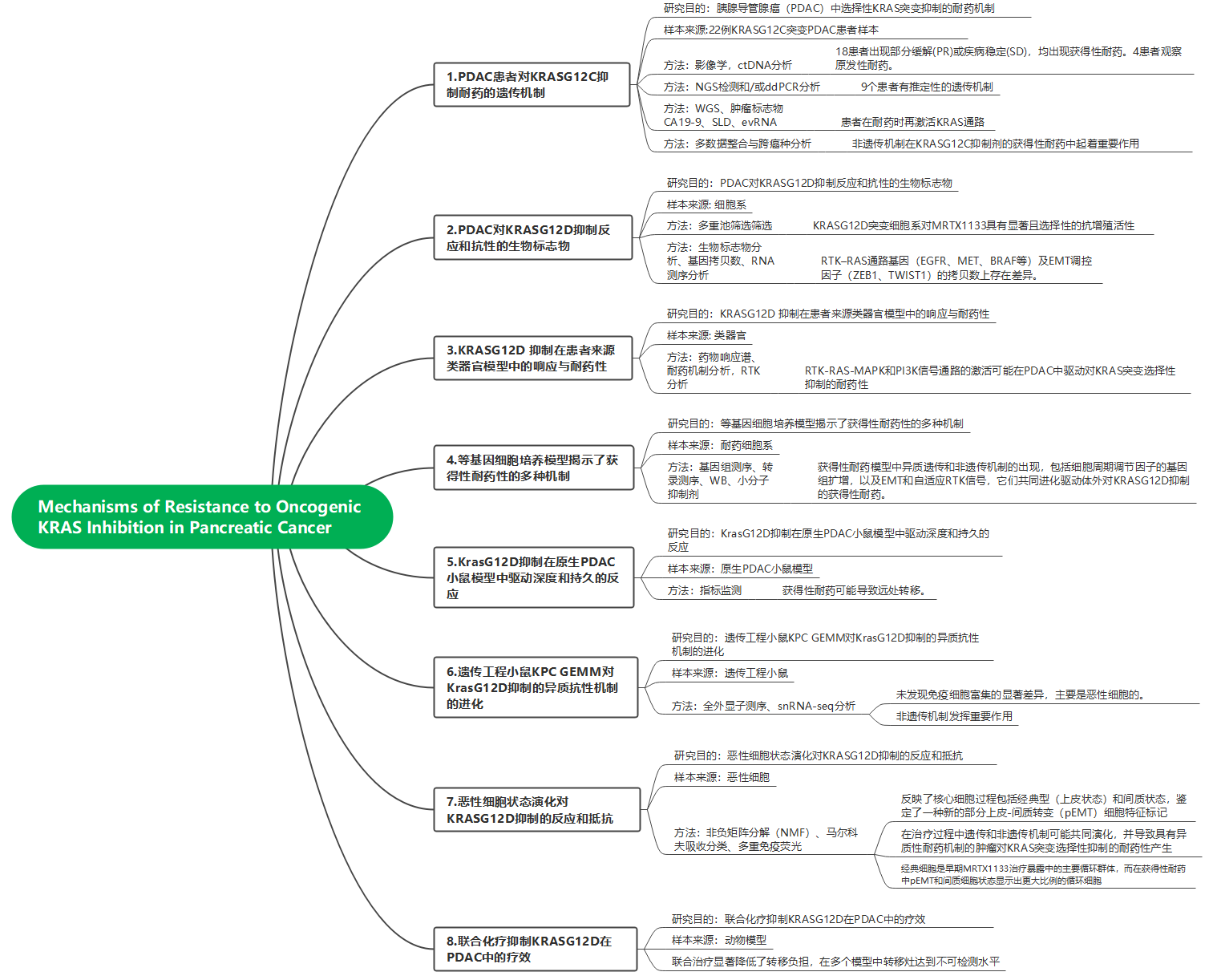
1. PDAC患者对KRASG12C抑制耐药的遗传机制
为了研究PDAC中对突变选择性KRAS抑制的耐药机制,对接受KRASG12C抑制剂治疗的KRASG12C突变型PDAC患者的治疗前(PT)和疾病进展后(EOT,即治疗结束时)血浆样本中的ctDNA进行了测序。结果表明:在这组患者中,22名PDAC患者中有18名(81%)显示部分缓解(PR)或疾病稳定(SD),但他们均发展出获得性耐药。而在22名患者中,有4名(患者1, 9, 10, 11)表现为原发性耐药,伴随疾病进展(PD)。通过NGS和/或ddPCR检测,KRASG12C等位基因在大多数PT样本(21/22)和EOT样本(20/22)中可被检测到(图1A和C)。在22名患者中,9名患者(41%)被鉴定出具有预测的原发性或获得性耐药的遗传机制(图1A-C)。在4名PD患者中,有3名的预测耐药机制包括MYC高水平扩增(患者1)、KRASG12C、BRAF、ERBB2和CDK4的联合增益/扩增(患者9)以及一个既存亚克隆KRASG12R变异的变异等位基因频率(VAF)增加(患者11)。在获得性耐药中,一些患者表现出治疗诱导的基因改变,包括KRASG12C的扩增/增益(患者6, 9, 13, 22),EGFR的扩增/增益(患者2, 13),PIK3CA的激活突变(患者13, 22),以及一个致癌性的NFE2L2D29H变异(患者21;图1C)。通过对患者4的EOT样本进行低覆盖率全基因组测序(WGS)分析,研究检测到多种治疗诱导的改变,包括ERBB2、MET、CDK6、MYC和ABCB1的高水平扩增(图1B和C)。此外,在EOT阶段(患者21)鉴定出一个治疗诱导的KRASA146P突变(图1A和C)。此前研究表明,这一突变在与G12C突变一起发生时,会导致对阿达格拉西布或索托拉西布类似物的耐药。在患者4和8中的治疗过程中对其血浆样本进行了动态采集,以观察他们对阿达格拉西布的动态响应和耐药过程。在治疗早期阶段,观察到循环肿瘤标志物CA19-9水平下降,并且两个患者的影像学肿瘤负荷相应减少。在获得性耐药和疾病进展阶段,检测到CA19-9的升高,同时SLD也增加。此外,治疗时发现在患者4的响应阶段中KRAS信号通路的转录活性降低,而在疾病进展阶段这一通路的活性增加,这表明患者在耐药阶段出现了通路再激活。
为了更好地定义KRASG12C抑制在不同癌症类型中的获得性耐药的预测遗传机制,研究整合了22名PDAC患者的数据(图1A-C),以及4名其他消化道癌症患者的新配对PT和EOT ctDNA谱。结果表明,ctDNA检测到的预测遗传耐药机制在51%的NSCLC患者、52%的CRC患者、46%的PDAC患者和75%的其他消化道癌症患者中被识别(图1D)。治疗诱导的KRAS改变(包括点突变和拷贝数扩增)在41%的CRC患者中观察到,但在NSCLC(21%)和PDAC(29%)中更少见。重要的是,在大比例的PDAC(54%)、NSCLC(49%)或CRC(48%)患者中,未能识别出预测的遗传耐药机制,这可能存在以下两种情况:ctDNA方法未能完全检测到治疗诱导的遗传驱动因子,或非遗传机制在KRASG12C抑制剂的获得性耐药中起到了重要作用。
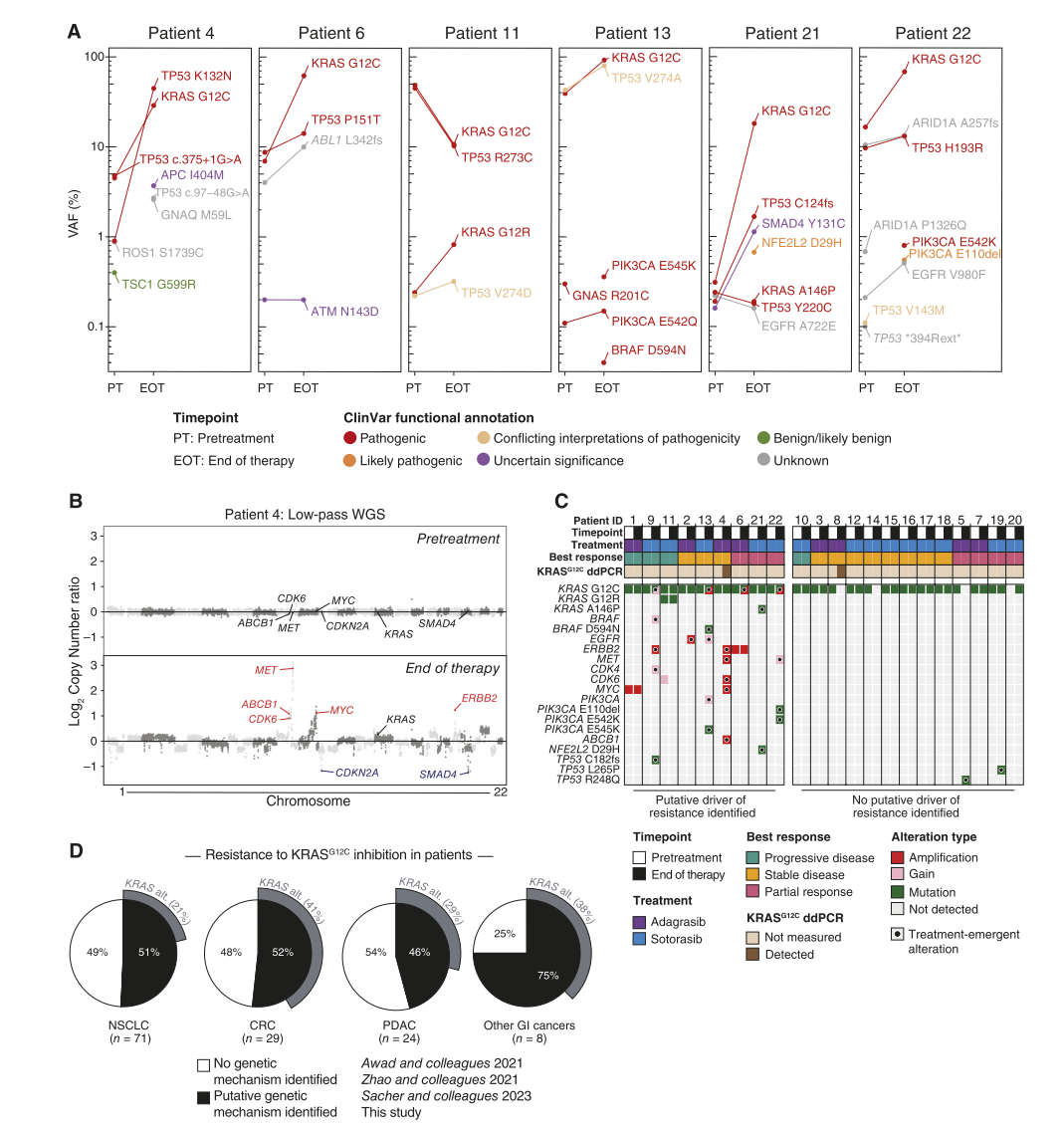
图1 基因改变与PDAC患者对KRASG12C抑制获得性耐药相关
2. PDAC对KRASG12D抑制产生反应和抗性的生物标志物
为了从更广泛的角度定义突变选择性KRAS抑制的响应和耐药潜在生物标志物,通过PRISM多重筛选法对877个人类癌症细胞系(涵盖不同的遗传和组织学背景)进行KRASG12D抑制剂MRTX1133的体外敏感性分析。KRASG12C突变在胰腺导管腺癌(PDAC)中非常罕见,并且仅开发了极少数KRASG12C突变型PDAC患者来源的模型,选择KRASG12D进行观察。研究观察到MRTX1133对KRASG12D突变细胞系表现出显著且选择性的抗增殖活性,而对KRAS野生型(KRASWT)细胞系或携带其他KRAS改变的细胞系的作用较弱(图2)。随后,根据MRTX1133的AUC将细胞系分类为“敏感型”或“耐药型”(图2B)。接下来,分析了KRASG12D突变PDAC细胞系(n = 16)中与MRTX1133敏感性或耐药性相关的生物标志物。未观察到与常见共突变的肿瘤抑制基因(如TP53、CDKN2A、SMAD4、ARID1A)改变相关的显著响应差异。此外,与敏感亚组相比,在耐药细胞系中观察到RTK-RAS通路基因(EGFR、MET、BRAF、ETV1)和上皮-间质转化(EMT)调控基因(ZEB1、TWIST1)的相对基因拷贝数升高(图2D)。在转录水平上,基因集富集分析(GSEA)显示,与敏感亚组相比,MRTX1133耐药细胞系中与EMT、细胞周期进程和MYC活性相关的基因上调(图2E)。体外PDAC细胞系模型中未观察到基于细胞状态(基底样/经典样轴)对MRTX1133的响应进行分层。在蛋白水平上,耐药细胞系中PI3K-AKT-mTOR信号通路效应蛋白(包括PI3K-p85α和p110α亚基、mTOR、Rictor、pS6 S235/S236)、翻译调控因子(elF4G、eEf2K)以及总和磷酸化的(pS445)BRAF蛋白水平较高,而MRTX1133敏感细胞系中E-Cadherin和β-连环蛋白(β-catenin)表达较高(图2F)。此外,在MRTX1133治疗后,上皮型敏感细胞系(HPAC和Panc 05.04)和间质型耐药细胞系(PANC-1和KP-4)中磷酸化ERK(pERK)水平均有所抑制,但间质细胞系在基线及MRTX1133治疗后显示出更强的PI3K-AKT-mTOR信号通路活性,这表现为磷酸化-S6(pS6 S235/S236)水平的增加。因此,PI3K-AKT-mTOR通路的差异激活可能在具有EMT特征的细胞中驱动了体外KRAS抑制的基线耐药性。
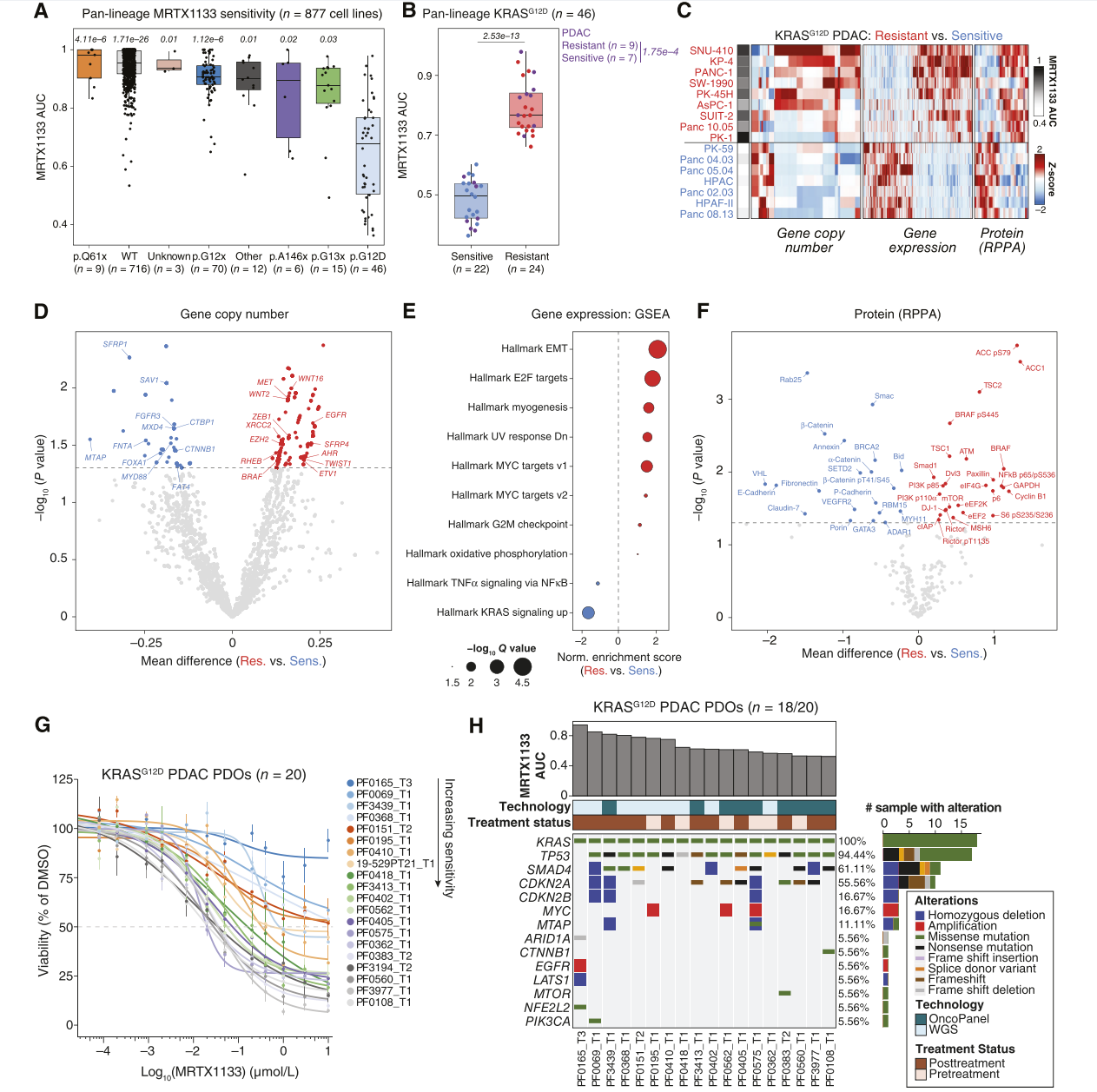
图2. 多组学分析确定了KRASG12D抑制反应和耐药的基线特征
3. KRASG12D抑制在患者来源类器官模型中的反应和耐药性
在来自初治和既往治疗过的癌症中的PDAC类器官(PDO)模型中,对MRTX1133的响应存在一个动态范围,从一些模型中发现明确的剂量依赖性敏感性到其他模型中的近乎完全耐药性(图2G)。正如在PRISM细胞系数据中观察到的那样,PDO对MRTX1133的响应并未因主要肿瘤抑制基因的改变而分层。然而,在最不敏感的PDO中注意到了其他潜在的耐药机制,例如EGFR扩增以及一个已知的NFE2L2D29H突变,该突变可破坏KEAP1结合,并已被证实可导致对MAPK靶向疗法的耐药性(图2H)。对MRTX1133耐药性第二强的PDO(PF0069_T1)被发现携带一个PIK3CAG118D突变,这是一个已知的致癌改变,会导致增强的PIK3CA活性和信号传导。相反,对MRTX1133表现出最大敏感性的PDO(PF0108_T1)携带一个CTNNB1T41A突变,这是一种已知的稳定化突变,驱动Wnt/β-catenin信号通路(图2H)。与细胞系中观察到的一样,研究中未在PDO模型中基于基底样/经典型转录特征的分类显著区分MRTX1133的响应。此外,研究发现去除标准类器官培养基中的EGF和FGF导致了所有研究模型的响应一致性增强,进一步确认了RTK-RAS通路刺激在驱动KRASG12D抑制基线耐药性中的作用。PDO的研究结果支持了RTK-RAS-MAPK和PI3K通路的激活可能驱动PDAC中对KRAS突变选择性抑制的耐药性。
4. 细胞培养模型揭示了获得性耐药性的多种机制
为了在体外模拟对MRTX1133的获得性耐药性,对敏感的小鼠细胞系(n=3)和人细胞系(n=2)模型进行MRTX1133剂量逐步增加处理,并获得了与各自的亲本细胞系相比,对KRASG12D抑制的体外敏感性显著降低的耐药模型(图3A和B)。这些耐药细胞系对MEK抑制剂曲美替尼也表现出敏感性下降,但对化疗药物吉西他滨没有表现出敏感性变化,这表明这些细胞系对其他RTK-RAS-MAPK通路抑制剂存在交叉耐药性,但对化疗药物没有耐药性。当将这些耐药性小鼠细胞系移植到C57BL/6小鼠的皮下时,它们同样表现出对MRTX1133的体内耐药性(图3C)。通过全外显子测序(WES),未观察到任何细胞系中KRAS或其他RTK-RAS-MAPK通路基因的获得性点突变。此外,在耐药性人细胞模型中也未观察到拷贝数改变。然而,在三种小鼠细胞系中的两种(6499C4.R和6419C5.R)中,研究发现在获得性耐药时染色体5上Cdk6和Abcb1a/Abcb1b的高水平基因扩增,这一现象与对阿达格拉西布耐药的人PDAC肿瘤中观察到的同源区域扩增一致(患者4;图1B和C)。这提示了通过CDK6介导的细胞周期失调以及ABCB1A/B介导的潜在药物外排可能是获得性耐药的机制。在转录水平上,这些获得性耐药模型表现出显著且反复的上调EMT(上皮-间质转化)过程和细胞周期进程,这与PRISM数据中观察到的情况相似(图2和3E)。这些发现得到了蛋白水平的验证,在耐药模型中间质标志物(ZEB1、VIM)表达增加,和/或E-Cadherin表达减少(图3F)。如先前在MRTX1133耐药性人细胞系KP-4和PANC-1中观察到的情况(这两种细胞系富含EMT特征,并在KRASG12D抑制下表现出持续的PI3K–AKT–mTOR信号通路活性)。还发现耐药模型相比各自的亲本细胞系,在基线和MRTX1133治疗后普遍表现出更高的PI3K/mTOR信号活性(图3F)。鉴于RTK依赖的信号传导在PDOs中介导了对MRTX1133的基线耐药性,以及在获得性耐药模型中KRASG12D抑制后持续的PI3K–AKT–mTOR信号活性,研究假设了RTK上调的不同模式可能驱动获得性耐药模型中MRTX1133敏感性的差异。结果表明,尽管在MRTX1133治疗5小时后,未观察到亲本与耐药模型之间的RTK活性明显差异,但在MRTX1133治疗48小时后,耐药模型相比匹配的亲本模型表现出HGFR、EGFR、RET、AXL、ERBB2、ERBB3和MSPR(由MST1R编码)活化水平的增加,这表明多个RTK的活化可能促进耐药表型的形成(图3G和H)。此外,靶向RTK信号的小分子抑制剂在亲本和耐药模型中均表现出协同作用,进一步表明RTK信号对获得性耐药的贡献,并可作为与KRAS抑制联合治疗的可行治疗靶点(图3I和J)。这些观察结果突出了获得性耐药模型中遗传和非遗传机制的异质性,包括细胞周期调节因子的基因组扩增、EMT过程的参与以及自适应RTK信号通路的共同进化,这些机制共同驱动了体外对KRASG12D抑制的获得性耐药性。
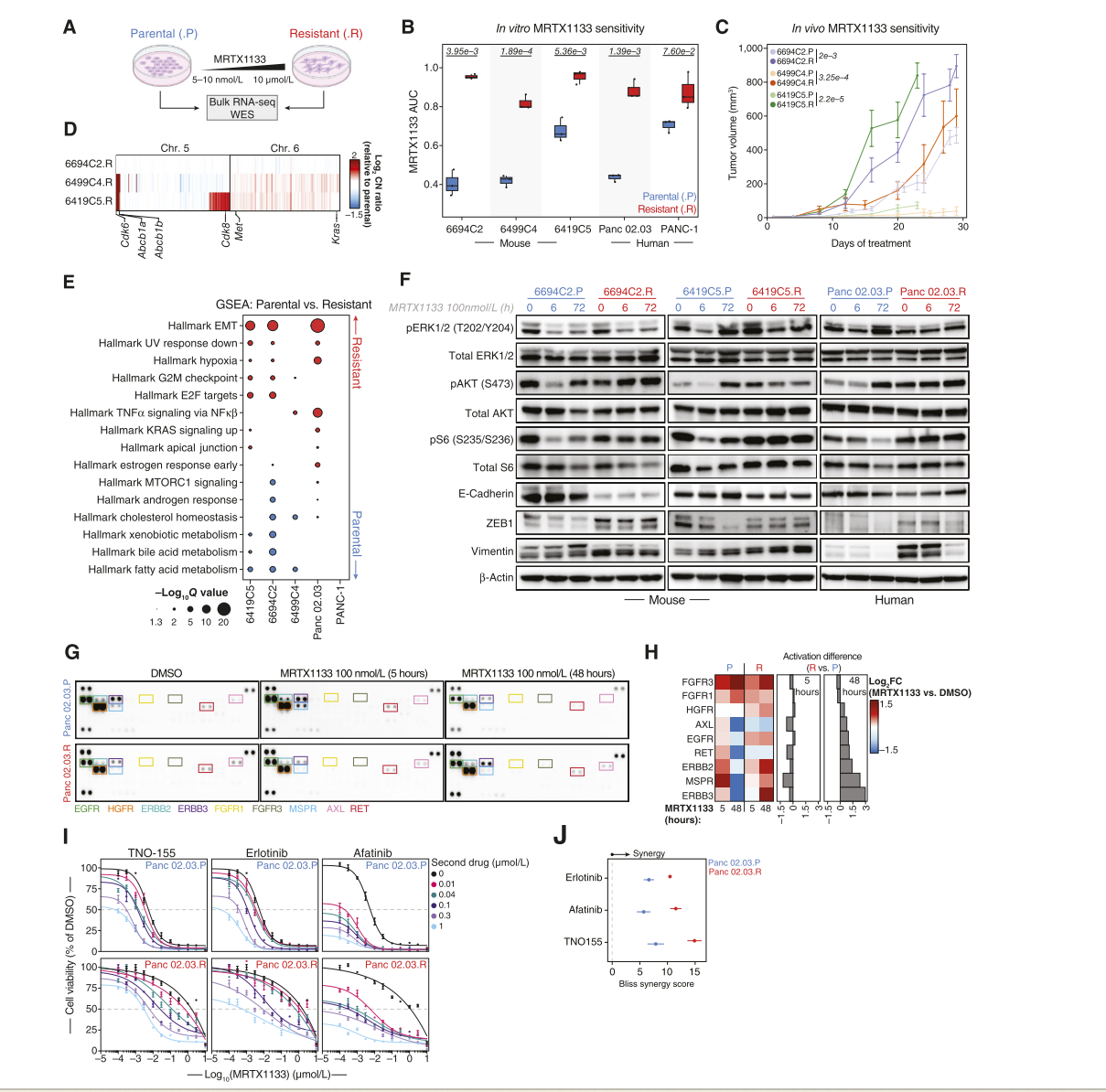
图3. CNAs, EMT和差异RTK信号驱动体外获得对KRASG12D抑制的抗性
KRASG12D抑制在原生PDAC小鼠模型中驱动深层和持久的反应
在体外评估了对MRTX1133的响应和耐药特性之后,进一步研究KRASG12D抑制剂在免疫功能完整的自发性KPC(KrasLSL-G12D/+; Trp53LSL-R172H/+; p48-Cre)胰腺导管腺癌(PDAC)遗传工程小鼠模型(GEMM)中的疗效。携带KPC基因型的小鼠通过触诊和超声连续监测肿瘤的生长,一旦肿瘤达到100到300 mm³的体积,每只小鼠随机分配到两个治疗组中的一个,分别接受载体对照(n=12)或MRTX1133(n=19)的治疗(图4A)。为了评估短期药理作用,4只MRTX1133治疗的小鼠(“早期MRTX1133组”)和3只对照组小鼠(“早期对照组”)在接受3天共6次治疗后被处理。MRTX1133治疗3天后测量的肿瘤体积表明,这些肿瘤在处理时对KRASG12D抑制有响应迹象。剩余的小鼠继续接受MRTX1133治疗(“终点MRTX1133组”,n=15)或对照治疗(“终点对照组”,n=9),直至达到生命终点,从而评估对MRTX1133的响应以及长期适应和耐药性。结果表明,MRTX1133治疗显著延长了PDAC小鼠的总体生存期,MRTX1133治疗组小鼠的中位生存期为10.7周,而对照组小鼠的生存期为1周(P = 2.08×10⁻⁶,图4B)。在终点MRTX1133治疗队列中,27%(4/15)的肿瘤出现部分缓解(PR)和中度肿瘤体积减少,在肿瘤体积随后经历了一个稳定(SD)的阶段后,最终在持续MRTX1133治疗下出现进展【图4C(中部)和D】。在1/15只动物中观察到了原发性耐药,其肿瘤增长动力学与对照组小鼠相似【图4C(顶部和中部)】。此外,MRTX1133治疗组中67%(10/15)的小鼠表现出完全缓解(CR),即在治疗的前两周内快速且完全的肿瘤消退,并通过超声证实未检测到任何可见肿瘤【图4C(底部)和D】。在这组小鼠中,完全且持久的缓解持续到获得性耐药发生。在其中两只经历完全且持久的原发性肿瘤消退直至生命终点的小鼠中,有一只在尸检中发现腹膜转移,表明获得性耐药也可能表现为远处转移。因此,在KPC小鼠模型中,KRASG12D抑制导致了显著的肿瘤反应,随后出现获得性耐药。
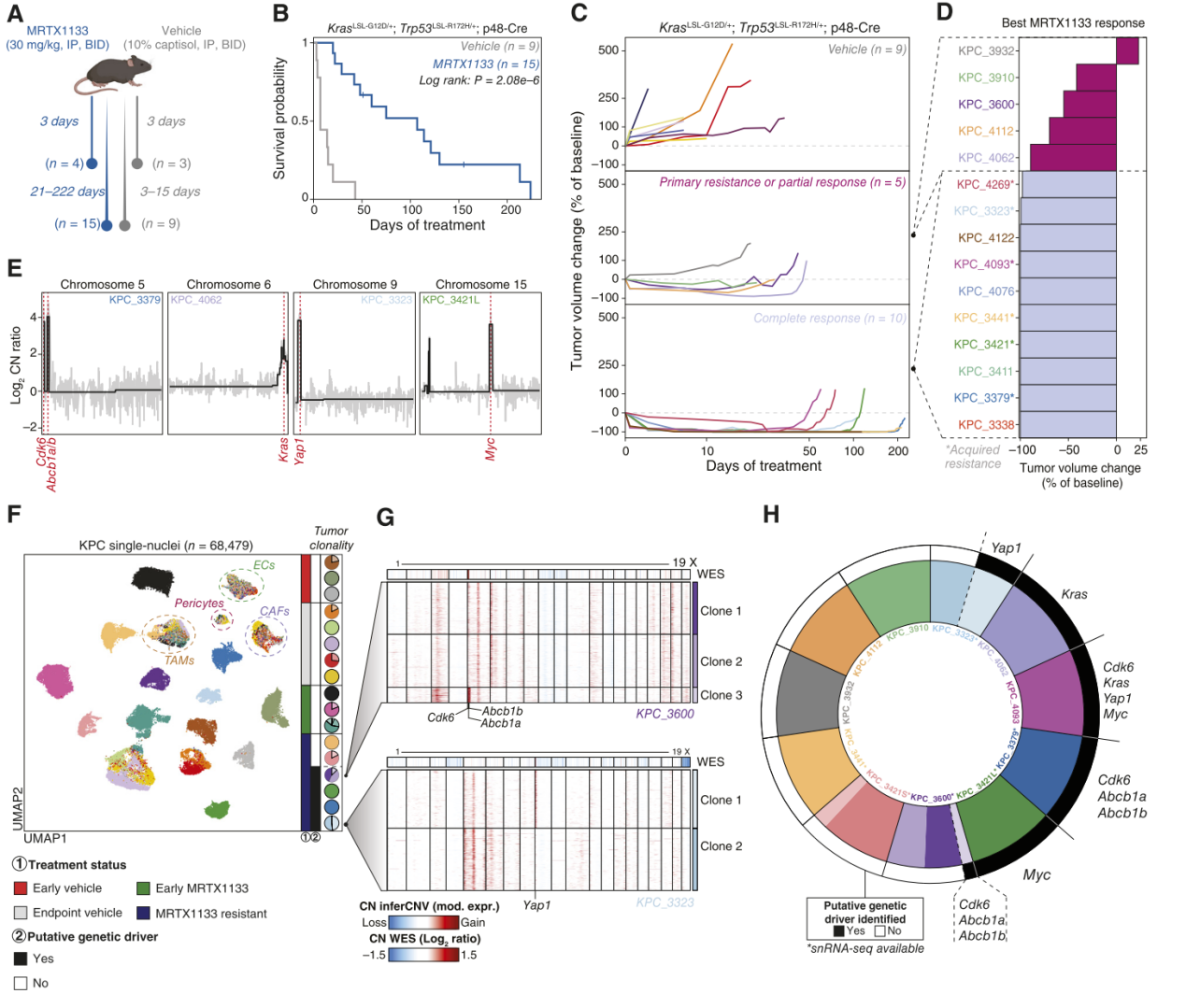
图4. 在PDAC的KPC小鼠模型中,异质基因组扩增驱动对KRASG12D抑制的抗性
6. KPC GEMM对KrasG12D抑制的异质抗性机制的进化
为了定义MRTX1133耐药的机制,研究者对接受对照或MRTX1133治疗的肿瘤进行了多模式表征,包括全外显子测序、单核RNA测序和免疫组化。在基因组层面,未观察到Kras中的药物结合位点突变。然而,在55%(6/11)的MRTX1133耐药终点肿瘤中观察到高水平的基因组扩增,而在对照组中未见。这些扩增包括2个肿瘤的Myc局部扩增、2个肿瘤的Kras扩增以及2个肿瘤的Yap1扩增(图4E)。此外,还在两例MRTX1133耐药KPC肿瘤中观察到包含Cdk6和Abcb1a/Abcb1b的染色体5着丝粒区域的高水平扩增,并在另一例肿瘤中观察到Cdk6单独扩增(图4E)。在一例MRTX1133耐药肿瘤中检测到多个不同的拷贝数变化(CNA),包括Kras、Cdk6、Yap1和Myc的扩增(KPC_4093)。为了进一步补充KPC小鼠中遗传耐药机制的研究,通过snRNA-seq评估了早期对照组、早期MRTX1133治疗组肿瘤,以及终点对照组和终点MRTX1133耐药组肿瘤的转录变化。从这些肿瘤中鉴定了五种不同的细胞群:癌相关成纤维细胞(CAF)、肿瘤相关巨噬细胞(TAM)、内皮细胞(EC)、周细胞和上皮细胞(图4F)。由于snRNA-seq数据中未检测到高质量的淋巴细胞群,通过流式细胞术进一步表征了肿瘤浸润的免疫细胞群。在终点对照组和MRTX1133耐药组肿瘤间未检测到显著差异的免疫细胞群。
进一步通过推断的CNA得分将每个肿瘤中的恶性上皮细胞与非恶性细胞群区分开来。接着,利用MAPK通路的转录活性作为KRASG12D抑制的药效学生物标志物,评估了恶性细胞核中的近端通路抑制效果。在MRTX1133治疗的肿瘤中,与对照组肿瘤相比,MAPK通路活性显著抑制。免疫组化检测也显示MRTX1133治疗的肿瘤中pERK蛋白水平降低,但在MRTX1133耐药组内存在异质性,表明在肿瘤获得KRASG12D抑制耐药后MAPK通路的激活存在差异。接着,利用单核推断的CNA谱分析了KPC队列中肿瘤内的遗传异质性。此外,研究发现WES和snRNA-seq检测到的基因组扩增之间具有一致性,其中后者揭示了多个肿瘤中存在的多亚克隆【图4F(右侧饼图)和G】。在部分MRTX1133耐药肿瘤中,WES检测的潜在耐药驱动因素在通过snRNA-seq推断的CNA中显示为亚克隆。例如,在KPC_3323中,观察到两个不同的克隆,其中Yap1扩增仅存在于克隆1,占该肿瘤恶性细胞的48%。在KPC_3600中,Cdk6/Abcb1a/Abcb1b扩增存在于一个次要克隆(克隆3),占该样本恶性细胞的13%。总体而言,在通过WES和/或snRNA-seq分析的11例肿瘤中,观察到6例细胞具有潜在的遗传耐药驱动因素,其中两例肿瘤显示这些改变具有亚克隆异质性(图4H)。正如患者数据(图1D)中先前所建议的,在KPC队列中的某些亚克隆或整个肿瘤样本中缺乏明显的耐药相关基因组改变,表明非遗传机制也可能在驱动KRASG12D抑制的获得性耐药中起重要作用。
7. 恶性细胞状态演化对KRASG12D抑制的反应和抵抗
为了识别KrasG12D抑制耐药的非遗传驱动因素,在snRNA-seq数据集中评估了肿瘤中恶性细胞的转录变异(图5A)。使用非负矩阵分解(NMF)方法,鉴定了核心细胞过程以及沿上皮–间质轴的细胞身份,包括经典(上皮)和间质细胞状态(图5A)。此外,发现了一种新型的部分EMT(pEMT)特征,与先前描述的上皮–间质轴上的混合状态类似。接着,通过一种基于Markov吸收的分类方法评估了核心细胞状态(经典、间质和pEMT元程序)之间的演化。细胞状态群体间的差异基因表达分析鉴定出了与经典状态(例如Tff1、Tff2、Muc5ac)和间质状态(例如Zeb1、Snail1、Vim)相关的特定基因,以及细胞状态特异性上调的标志性细胞过程和功能(图5B和C)。间质细胞中观察到EMT特征和高KRAS信号,而经典细胞群体富集了细胞周期过程和增强的外源性代谢活动。pEMT细胞状态的特征包括细胞应激、增强的翻译、蛋白质稳态、氧化磷酸化和Myc信号(图5C),其中一些特征先前已被证明与KRAS通路抑制或抑制耐药性相关。最后,通过基因调控网络分析鉴定出了与肿瘤中元程序表达相关的主要转录调控因子(例如Zeb1调控间质状态,Spdef和Creb3l1调控经典状态)(图5D)。此外,还发现了新的可能调控pEMT细胞状态的因子,包括Ybx1、Sox4以及AP1复合体的组成部分(例如Jun、Junb和Jund)。接下来,研究发现来自对照组的KPC小鼠的肿瘤显示了沿经典-间质轴的混合表型群体。在MRTX1133治疗的肿瘤中,观察到两种细胞状态承诺的不同模式。在早期(3天)MRTX1133治疗队列的肿瘤中,观察到间质型PDAC细胞的显著耗竭和经典细胞状态的富集,这表明间质型PDAC细胞可能比经典型PDAC细胞对KrasG12D抑制更为敏感。此外,在MRTX1133耐药肿瘤中,通过snRNA-seq观察到向pEMT(耐药肿瘤中60%的细胞核)和间质(耐药肿瘤中30%的细胞核)恶性细胞状态的显著转变(图5E)。对KPC肿瘤的组织学分析显示,MRTX1133耐药肿瘤中未分化肿瘤细胞的强烈富集。此外,在MRTX1133治疗的肿瘤中,沿经典-pEMT-间质轴的循环细胞分布发生了转变,经典细胞是早期MRTX1133治疗暴露中的主要群体,而在获得性耐药中pEMT和间质细胞状态更为显著。
鉴于KPC模型中间质型PDAC对KRAS抑制的相对敏感性,进一步寻找人类PDAC转录亚型与MRTX1133体内响应相关的证据。人源异种移植(PDX)模型已被证明能够准确再现人类PDAC亚型的多样性,包括经典型和基底样亚型,其中基底样PDAC表型与PDAC中的间质细胞状态表现出表型相似性。因此,研究者分析了一组KRASG12D突变PDX模型,这些模型最近被报道在MRTX1133治疗下表现出部分缓解(PR)或疾病稳定(SD)。在这些PDX模型中,研究者观察到在所有基底样模型中,MRTX1133均导致显著的肿瘤生长抑制和部分缓解(PR)。相反,在经典模型中,主要观察到疾病稳定(SD),3/4模型几乎未见肿瘤回归(图5G)。此外,与对照处理相比,在KRAS抑制剂治疗3天后,3/7个PDX模型中的基底样特征显著减少(图5G)。这些体内PDX结果支持了基底样PDAC相比经典PDAC对KRAS抑制的相对响应增强。
为了研究遗传和非遗传驱动因素对KRASG12D抑制耐药的相互作用,整合了先前定义的遗传亚克隆(图4G和H),并调查了单个MRTX1133耐药肿瘤中pEMT耐药元程序的分布(图5H)。在4个检测到克隆性或亚克隆性潜在遗传耐药驱动因素的肿瘤中,有3个肿瘤表现出相对较低的pEMT特征表达。例如,携带Cdk6/Abcb1a/Abcb1b扩增的恶性细胞核,无论它们来自基因组异质性肿瘤中具有扩增的亚克隆(KPC_3600,克隆3),还是来自具有克隆性扩增的肿瘤(KPC_3379),其pEMT元程序均表现出相对较低的富集。同样,来自KPC_3323的Yap1扩增亚克隆(克隆1)相比于同一肿瘤中未扩增Yap1的克隆2,其pEMT特征也相对较低。相反,其他肿瘤在耐药时同时表现出遗传驱动因素和pEMT表型。例如,KPC_3421L表现出克隆性Myc扩增和相对高的pEMT富集。而KPC_3441虽然未发现任何遗传耐药驱动因素,但却表现出较高的pEMT细胞状态表达(图5H)。因此,pEMT程序与潜在遗传驱动因素的共同出现存在异质性,这表明pEMT状态可能在某些肿瘤中作为一种独特的耐药路径,或在其他肿瘤中与遗传驱动因素共同发挥作用。综上所述,在治疗过程中遗传和非遗传机制可能共同演化,并导致具有异质性耐药机制的肿瘤对KRAS突变选择性抑制的耐药性产生。
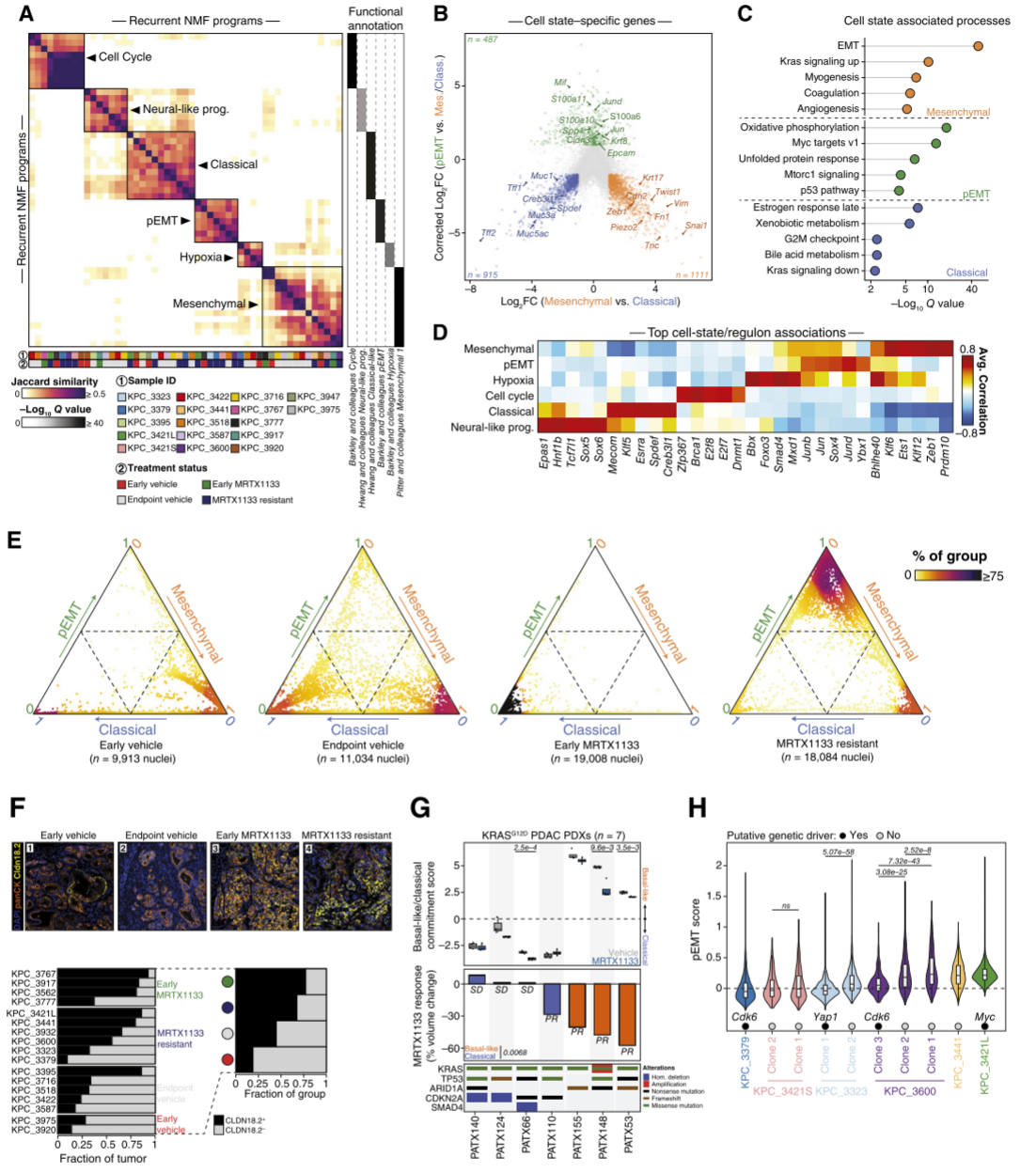
图5. 在活体内的MRTX1133应答和耐药期间沿上皮-间质轴的恶性细胞状态演变。
8. 联合化疗和抑制KRASG12D在PDAC中的疗效
患者数据表明,与预后较差的基底样PDAC亚型相比,经典型PDAC亚型对化疗的响应更好。因此,探讨了联合化疗是否可以显著提高Kras抑制在多种原发性和转移性PDAC小鼠模型中的疗效。将6694C2小鼠PDAC细胞正位植入C57BL6/J小鼠的胰腺中,当通过超声检测到肿瘤后,将小鼠随机分为对照组、吉西他滨/(n)白蛋白结合型紫杉醇(GnP)组、MRTX1133单药组或联合治疗组(MRTX1133/GnP)。GnP治疗与对照处理的动物模型相比仅引起适度的肿瘤生长抑制(图6A)。MRTX1133单药治疗导致肿瘤回缩,但在治疗两周后肿瘤复发。与以往报道一致,短期MRTX1133治疗使得CD4+和CD8+ T细胞在此模型中增加。联合MRTX1133和GnP治疗也导致肿瘤回缩,显著防止了肿瘤复发,且显著延长了肿瘤生长抑制时间(图6A)。这些数据表明,联合化疗和KRASG12D抑制显著提高了原发性肿瘤的生长控制,相比单一治疗效果更佳。
大多数PDAC患者在确诊时已发生转移,而KRAS抑制剂的早期临床试验主要在化疗作为当前护理标准的这类患者中进行。因此,需要进一步探讨了单一MRTX1133和联合化疗治疗在一种新型PDAC转移模型中的疗效。检测了中期转移性PDAC细胞模型(6694C2-LM)皮下肿瘤中免疫细胞的浸润情况,比较了对照和MRTX1133治疗组。与先前的研究一致,即致癌KRAS驱动GM-CSF生成并招募粒细胞,发现MRTX1133治疗导致免疫抑制性生长因子减少,以及粒细胞和单核细胞来源的髓样抑制细胞浸润减少。在这一低免疫原性模型中未观察到T细胞浸润的变化。在6694C2-LM转移模型中,进一步评估了辅助MRTX1133、GnP或二者联合治疗对转移扩散的控制能力。小鼠接种皮下肿瘤后,在所有组中肿瘤达到相似大小时进行手术切除(图6E)。术后小鼠随机分组,分别接受对照、MRTX1133、GnP或MRTX1133/GnP联合治疗8周,然后牺牲小鼠并收集器官以检测转移情况。结果显示,仅接受辅助性对照、MRTX1133或GnP治疗的小鼠均出现大量转移(图6F)。然而,MRTX1133与GnP联合治疗将转移负担减少至不可检测的水平。最后,通过静脉尾注模型评估这些治疗方案对已建立肺转移的疗效。与载体处理的小鼠相比,GnP治疗减少了转移的总数量(图6H)。与GnP单药相比,单一MRTX1133显著减少了总转移负担。在两种情况下,MRTX1133和GnP的联合治疗显著优于其他所有治疗条件,参与实验的小鼠肺部均未检测到转移灶(图6H)。这些结果表明,在一种侵袭性转移性PDAC模型中,结合突变选择性Kras抑制剂的联合化疗具有显著优势。
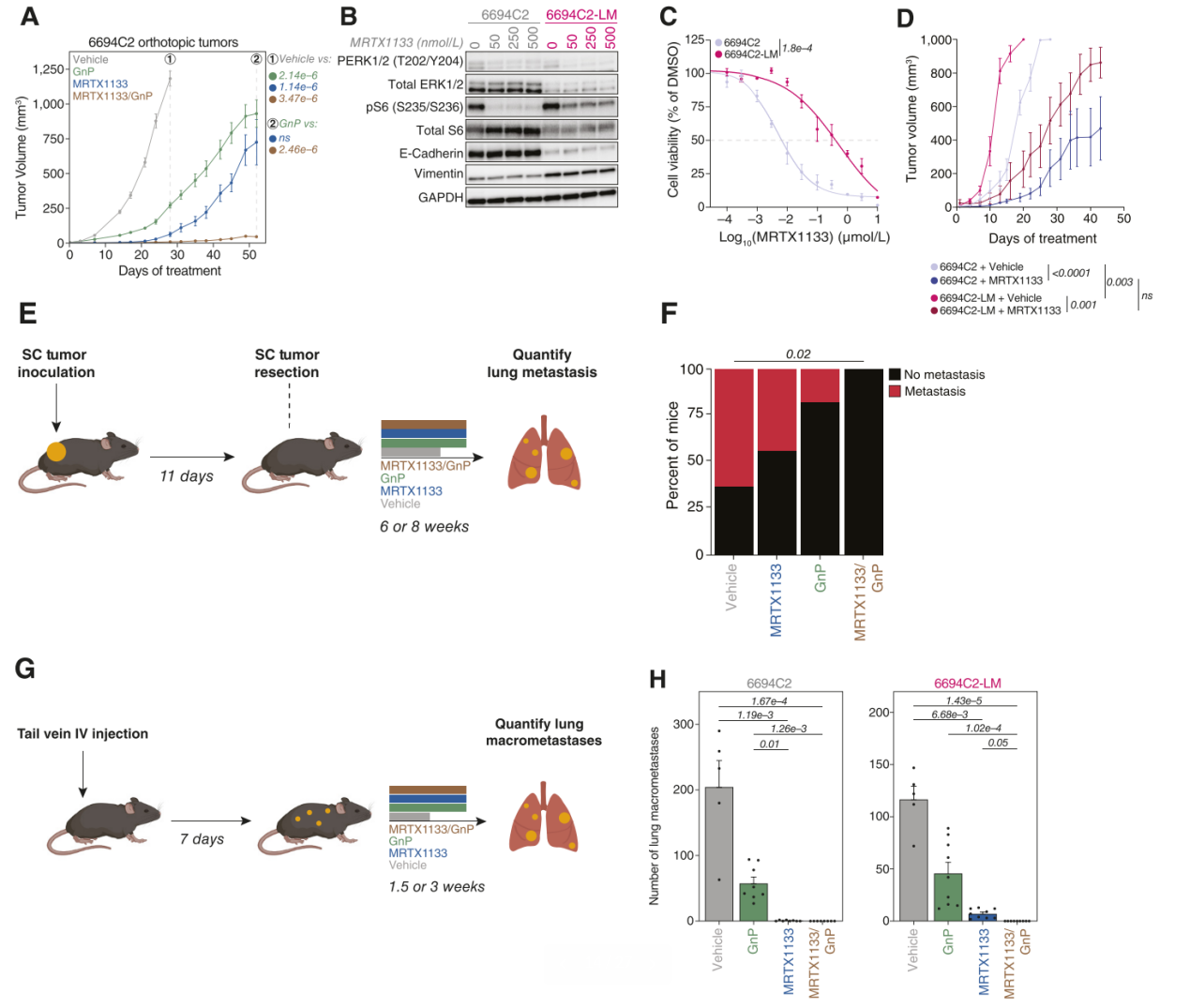
图6. KrasG12D抑制和化疗联合治疗原发性和转移性PDAC的疗效

汇报人:邹宇豪
导师:刘世喜
审核:邱轲、谢尔杰、任建君
