


华西耳鼻喉学术前沿速递——文献精读(第51期)
精读分享│【Nature Communications】:蛋白质组学分析揭示RNA是肿瘤新抗原的重要来源
英文题目:Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification
中文题目:蛋白质组学分析揭示RNA是肿瘤新抗原的重要来源
期刊:Nature Communication(IF=14.7)
单位:德国癌症协会(DKTK),慕尼黑工业大学,TUM医学院,转化性癌症研究中心,慕尼黑,德国
发表时间:2023年8月



摘要
系统性泛癌分析能够揭示癌症免疫原性和患者生存有关的共同特征的重要性。本研究中纳入25 种肿瘤类型的32名患者全面多组学数据,探究基于蛋白质组学的新抗原。通过使用优化的算法,研究发现了大量肿瘤特异的和肿瘤相关的抗原。为了构建识别队列中新抗原的分析流程,研究将DNA和RNA测序与基于MS的肿瘤样本免疫肽组学相结合,评估其免疫原性并进行深入验证。研究在大多数表现出部分免疫原性的患者中检测到多种非典型的HLA结合肽;在验证流程中鉴定出32种潜在的新抗原候选物。大多数新抗原候选物源自RNA数据集中识别的变异,说明RNA作为癌症抗原的尚未得到充分研究的来源相关性。这项研究强调了以RNA为中心的变异检测对于识别共享生物标志物和潜在相关的新抗原候选物的重要性。
引言
基因变异不仅是癌症发生发展的核心因素,还会导致新抗原的形成,这些新抗原有可能引发抗肿瘤的免疫反应。新抗原可以被作为外来物质识别,并成为新抗原特异性T细胞的靶点。因此,识别这些新抗原对于开发新型免疫疗法至关重要。然而,大多数新抗原在癌症患者之间并不共享,而通过计算预测出的新抗原候选物的数量通常非常庞大,临床验证这些候选物的可行性有限。
为此,研究团队之前提出了一种基于蛋白质组学的策略,将免疫沉淀的HLA-I类(pHLA-I)肽段的液相色谱-串联质谱(LC-MS/MS)与黑色素瘤肿瘤的全外显子组测序(WES)相结合,以在蛋白质水平上识别和验证新抗原。该方法在实体肿瘤中得到了验证,但识别出的新抗原数量有限,且需要在不同的癌症类型中进一步验证。
研究还表明,不仅编码外显子中的体细胞突变可以产生新抗原,非编码区转录本、内含子区域和剪接位点也可能成为新抗原的来源。此外,RNA编辑等RNA变异事件也受到了越来越多的关注。RNA编辑是一种广泛的转录后机制,在正常细胞中会引发特定的、可重复的核苷酸变化,但在癌症中常发生异常,导致癌症蛋白质组的多样化。因此,研究中进一步深入探讨了RNA编辑产生的变异作为异常表达肽段的来源。
由于RNA调控受顺式和反式调控元件的影响,而这些元件常被体细胞突变或致癌信号通路所干扰,因此癌症相关的RNA编辑产生的抗原可能部分是真正的新抗原,因而对癌症免疫疗法具有高度研究价值。因此,研究在全外显子组测序之外,还纳入了肿瘤转录组学,以检测由RNA变异事件产生的新抗原。
此外,研究团队此前已表明,在新抗原识别过程中,整合光谱预测特征到MS光谱匹配过程中的重新评分方法可以有效处理更大的搜索空间,并提高分析的覆盖率和敏感性。因此,研究使用人工智能算法Prosit,并利用基于Prosit的重新评分工作流程来进行新抗原识别。
在这项工作中,研究团队利用了MASTER队列,纳入32名患有不同类型肿瘤的患者,测试了改进的蛋白质组学流程。在大多数患者中,研究团队识别到的新抗原主要来源于RNA变异。研究还通过T细胞表型分析表明,新抗原的免疫原性与T细胞浸润的增加相关。因此,该数据表明,基于蛋白质组学的新抗原识别在泛癌队列中是可行的,并且RNA来源的新抗原可能是开发免疫疗法的高度相关的靶点。
研究结果思维导图
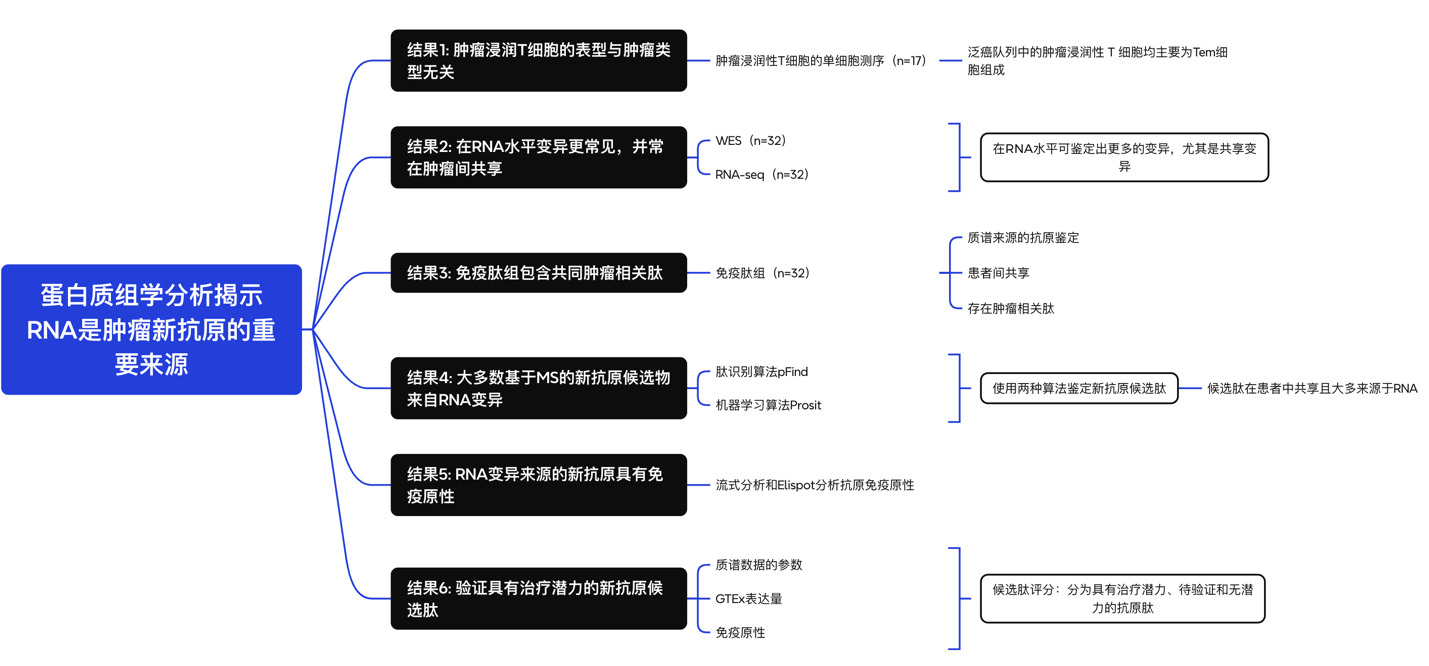
研究主要内容
为了在ImmuNEO MASTER队列中识别常见的泛癌免疫相关特征和新抗原候选物,研究创建了一个用于分析肿瘤样本的通用工作流程,如图1所示。首先,通过流式细胞术从肿瘤组织的肿瘤微环境(TME)中分选出肿瘤浸润T细胞(图 1a)。接下来,使用队列中肿瘤样本的全外显子测序(WES)/全基因组测序(WGS)和RNA测序(RNA-seq)数据进行分析(图 1b)。紧接着,对 pHLA-I 进行了免疫沉淀,随后进行了 MS 分析,以鉴定抗原呈递的免疫肽组(图 1c)。然后,使用 pFind 将基因组和转录组数据与基于 MS 的免疫肽组学数据相结合(图 1d)。作为关键创新,研究纳入了 RNA 测序数据并使用人工智能算法 Prosit 来提高研究的新抗原发现流程的覆盖率和灵敏度。并且使用患者来源的自体或健康供体(HD)来源的同种异体匹配 T细胞在体外评估已鉴定的新抗原候选物的免疫原性(图 1e)。重要的是,研究通过验证和评估其在正常组织表达数据中的普遍性来验证研究优化的流程中确定的新抗原候选物(图 1f)。最后,为了揭示新抗原识别的潜在临床条件(这可能是临床应用的关键知识),研究将已验证的总和免疫原性新抗原的数量与 TME 免疫表型数据相关联。
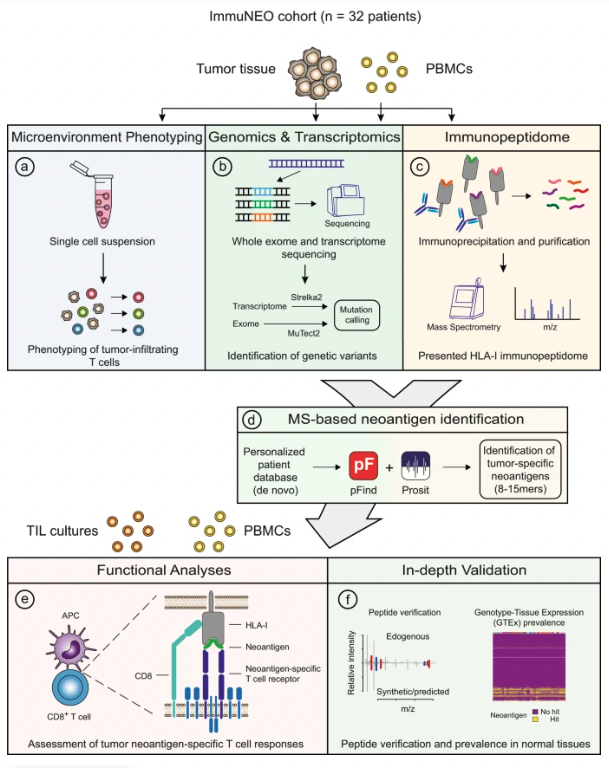
图1
1. 肿瘤浸润 T 细胞的表型与肿瘤类型无关
为了研究能否将肿瘤微环境中观察到独立于肿瘤类型的免疫学特征并将其与临床结果联系起来,研究对新鲜肿瘤组织进行了流式细胞免疫表型分析。在 17 名有足够肿瘤组织的患者中,对 T 细胞亚群进行了探究。
首先,研究观察了每种肿瘤中CD8+ T细胞的相对细胞数量(图 2a)。两例黑色素瘤和一例有错配修复缺陷 (dMMR) (ImmuNEO-11 T2) 的胰腺癌转移显示有大量 T 细胞浸润,与这些恶性肿瘤中经常出现的高突变负担相匹配。然而,其他肿瘤实体,包括肉瘤标本 (ImmuNEO-5),也存在大量肿瘤浸润淋巴细胞 (TIL) (图 2a )。无论肿瘤实体如何, CD8+细胞主要由效应记忆T(Tem; CD45RA- CD62L low ) 细胞组成(图 2b)。此外,在患者的不同转移瘤之间,CD8+ T 细胞亚群的分布与它们的解剖转移位置无关(图2b),尽管它们的相对细胞数量存在差异(图 2a)。由于TIL的功能状态与其潜在的抗肿瘤活性有关,研究分析了选定的活化标志物(HLA-DR 和 CD103)和抑制标志物(PD-1、TIM-3 和 LAG-3)的表达。为了解释总体细胞数量的差异并研究群体水平的活化状态,分别研究了表达至少一种标志物的 CD8+细胞上活化或抑制标志物的比例。不同肿瘤实体之间具有活化标志的CD8+ T细胞占比没有差异,并且在癌,肉瘤和黑色素瘤患者中存在高频率的具有抑制特征的肿瘤标本(图 2c)。
为了在队列中识别出临床相关的转录T细胞特征,研究对8名患者的分类CD8+ TIL进行了RNA测序。根据患者自肿瘤切除后的生存数据,将患者分为短期生存组(少于1年)和长期生存组(超过1年)。通过使用基因集富集分析(GSEA),研究者证明,与T细胞介导的细胞毒功能相关的通路在长期生存组中上调,而与一般炎症反应相关的通路在短期生存组中上调(图 2d)。此外,为了识别与生存相关的组织不可知特征,通过对数秩检验和Cox比例风险模型评估了每个参数对患者自肿瘤切除以来生存的影响(图 2e)。尽管CD8+ T细胞的数量和频率与生存率增加呈不显著的正相关趋势,但TME 中无抑制标志物的CD8+ T细胞的总体频率与生存率增加呈正相关(图2e)。此外,CD8+ Teff 亚群中无活化或抑制标志物的细胞频率也与生存率增加呈正相关,因此,该亚群中具有活化或抑制标志物的细胞比例升高与生存率降低呈正相关(图2e)。
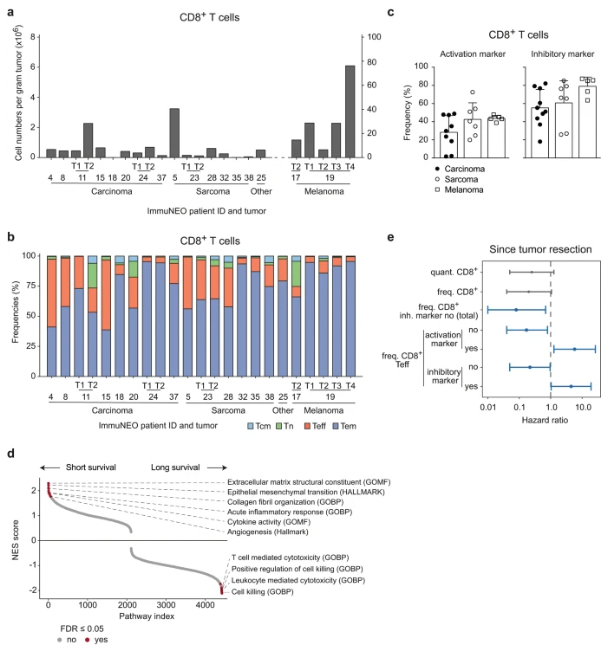
图2
总之,研究观察到,异质性泛癌队列中的肿瘤浸润性 T 细胞主要由独立于肿瘤实体的 Tem 细胞组成。此外,研究可以重现之前在同质性肿瘤队列中观察到的发现,例如在以高突变负荷为特征的恶性肿瘤中 TIL 数量增加,并观察到与该队列中的临床结果相关的CD8 + T 细胞中的特定转录通路。
2.在 RNA 水平变异更常见,并常在肿瘤间共享
研究在DNA和RNA水平上评估了肿瘤中变异的数量。由于这些数据是识别新抗原候选物的基础,稍后将通过基于MS的肿瘤免疫肽组分析进行交叉验证(图 1)。
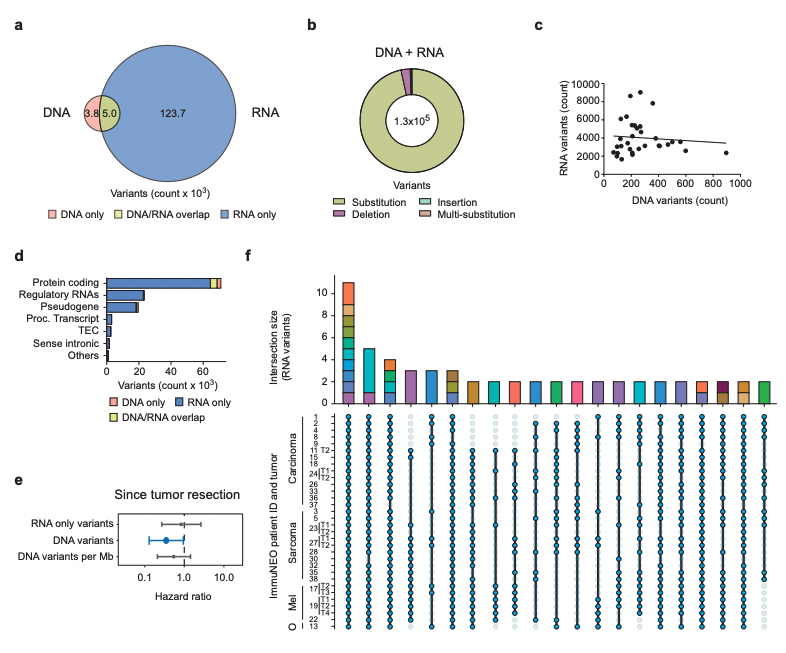
在泛癌症队列中,患者之间DNA和RNA变异的数量差异很大,但在不同肿瘤实体之间没有明显差异(图 3a)。平均而言,研究在每个肿瘤中发现了 302 个体细胞突变,但在RNA水平上发现了更多的变异,平均每个肿瘤有 4024 个变异(图 3a)。值得注意的是,大多数 DNA 变异也在 RNA 水平上发现(补充图 5a),凸显了 RNA 作为发现遗传变异来源的强大功能。总体而言,单核苷酸替换占 DNA 和 RNA 水平上发现的大多数变异,但对于一些变异也观察到了缺失和插入以及多核苷酸替换(补充图 5b)。有趣的是,每种肿瘤中鉴定出的 DNA 和 RNA 变异的数量之间没有相关性(补充图 5c),这表明体细胞突变水平低的肿瘤仍然可以含有大量的 RNA 变异。
与DNA水平相比,在RNA水平上检测到的变异数量更多,且非编码来源仍然不能解释这种显著的差异。RNA编辑事件可以为RNA变异提供额外的来源解释。为此,研究分析了DNA水平上相应基因座的覆盖率和所有仅在RNA水平上鉴定的变异的核苷酸交换模式。事实上,对于大多数RNA变异,可以在DNA水平上检测到相应的规范序列(图 3b),这表明这些变异的一部分可能来自RNA编辑事件,其中,大多数为腺苷(A)到鸟苷(G)的核苷酸交换(图 3c)。研究观察到DNA和RNA变体主要由错义变体组成,但RNA变体由更多的剪接位点和内含子变体组成(图 3d)。
此外,研究对该泛癌群体中共享的DNA和RNA变异特别感兴趣,因为这些变异可能导致潜在的共同新抗原,而这些新抗原可能成为免疫疗法的有吸引力的靶点。因此,研究调查了每个变异体被检测到的患者数量。正如预期的那样,绝大多数变异在 DNA 和 RNA 水平上都是独一无二的(图 3e、f)。事实上,在队列中,~97% 的变异在 DNA 水平上是独一无二的(图 3e),但在 RNA 水平上只有 89% 是独一无二的(图 3f)。再加上研究检测到的 RNA 变异比 DNA 变异多大约 10 倍,这意味着研究可以在 RNA 水平上识别出大约 37 倍更多的共享变异(在至少 2 名患者中检测到)。
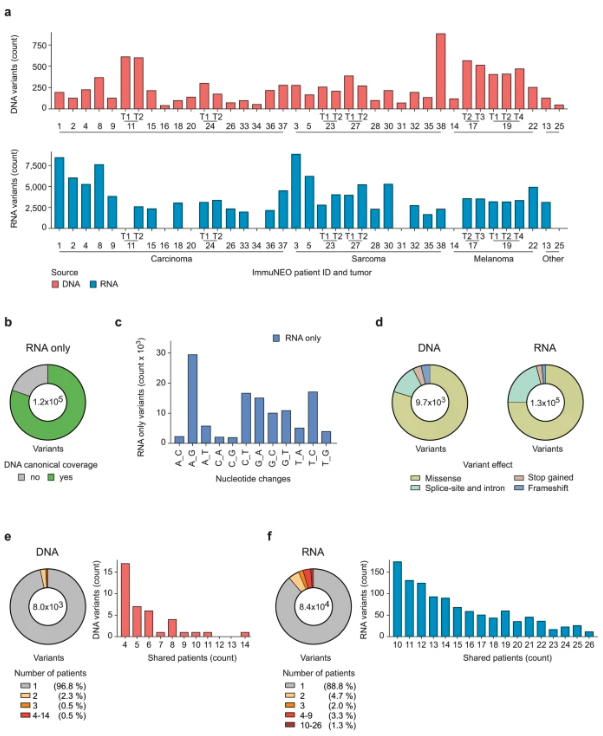
图3
为了阐明这些共享 RNA 变异是否在同一组患者中相互重叠,重点研究了至少在十个肿瘤标本中发现的 RNA 变体,这些变异至少有两种共享 RNA 变异(补充图 5f)。重叠的共享 RNA 变异不仅常见于肿瘤转移中,而且也存在于泛癌队列的不同肿瘤样本中(补充图 5f )。虽然发现这些组中的大多数共享 RNA 变异是独有的,但研究能够识别出 59个显示出一定程度重叠的共享变异。其中,11 种 RNA 变异存在于泛癌队列的所有患者和肿瘤转移中。
补充图5
总的来说,研究在RNA水平上鉴定出了更多的变异,特别是共享变异,并且相当一部分额外的 RNA 变异可能源自 RNA 编辑事件。
3. 免疫肽组包含共同肿瘤相关肽
为了表征泛癌队列中的肿瘤免疫肽组,研究对pHLA-I进行了免疫沉淀,然后进行了MS分析。与 DNA 和 RNA 变异的数量相似,肽的总体数量在患者之间差异很大,不同肿瘤实体之间没有明显偏差(图 4a)。平均每个肿瘤可鉴定出约 5075 种肽(图 4a),长度为 8 至 15 个氨基酸。
通过关注人类蛋白质图谱中描述的癌症相关基因衍生肽,研究发现 36% 的此类肽在患者之间是共享的(图 4b),而且多达 18 名患者中存在相当数量的此类肽(图 4c)。此外,研究使用 CTpedia 数据库分析了已报道的癌症睾丸抗原 (CTA) 衍生肽,并在队列中发现了大量 CTA 肽(图 4d)。虽然大多数 CTA 肽仅在一名患者身上发现独有,但研究鉴定了多种源自 CTA 相关基因的肽,这些肽存在于相当一部分患者中,与肿瘤类型无关(例如ATAD2、SPAG9、ODF2和KIAA0100)(图 4d)。
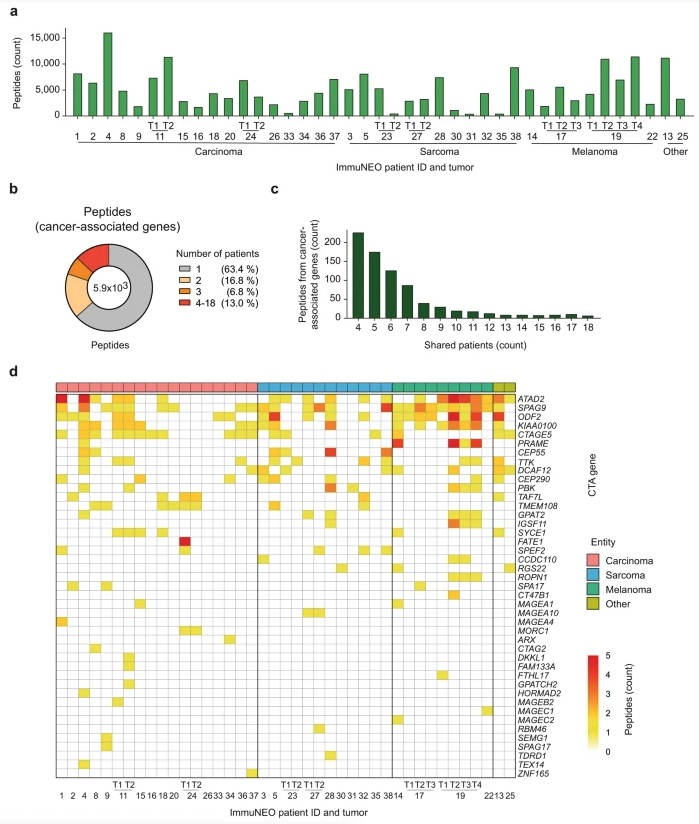
图4
因此,对泛癌队列中的免疫肽组进行研究,发现了许多可用于免疫治疗的潜在肿瘤相关抗原候选物。
4. 大多数基于MS的新抗原候选物来自RNA变异
为了识别新抗原候选物,研究使用肽识别算法pFind,随后通过机器学习算法Prosit重新评分(图 1),可以将已识别的新抗原候选物的总数增加(图 5a)。
通过该蛋白质组学流程,研究在 24 名不同肿瘤实体的患者中鉴定出 90 种新抗原候选物(占所有患者的 75% 和具有 RNA 测序数据的患者的 88%),每名患者鉴定出 1 至 13 种新抗原候选物(图 5b),这表明大多数癌症患者都蕴藏着个性化免疫治疗的潜在靶点。研究者没有观察到患者之间共享的新抗原候选物,但是,一名黑色素瘤患者的两个转移瘤(ImmuNEO-19)之间共享三种肽,一名 dMMR 患者的两个不同肿瘤样本(ImmuNEO-11)之间共享一种肽。有趣的是,研究者在两例患者(ImmuNEO-4和 −23)中发现了两种新抗原候选物,它们分别来自MAP4K5(IN_04_F,1.5% FDR;32个肿瘤样本间共享)和AC024075.2(IN_23_A,4.3% FDR,24个肿瘤样本间共享)中的共享变异。由于这两种共享变异均能产生至少在一名患者中存在的pHLA-I,因此这两种肽有可能存在于具有这些变异的其他患者中,但由于患者免疫肽组的检测限制而被遗漏。
所有已鉴定的新抗原候选物的肽长度范围为 8 至 14 个氨基酸(图 5c)。在 90 种已鉴定的新抗原候选物中,79 种仅来自 RNA 变异,而只有三种仅来自 DNA 变异,八种是两种来源共享的(图 5d)。与 RNA 变异的总数相比,研究者可以在 DNA 水平上检测到大多数已鉴定的新抗原候选物的相应序列,这些新抗原候选物完全来自 RNA 变异(图 5e)。此外,这些变异中的许多还包含与 RNA 编辑相关的 A 到 G 核苷酸交换模式(图 5f)。这表明 RNA 改变机制(如RNA编辑)可能是新抗原形成的重要来源。关于产生新抗原候选物的变异的变异效应,错义变异仍然最为丰富,然而,与总体检测到的变异相比,剪接位点和内含子变异更为普遍(图 5g,左)。大多数新抗原候选物来自蛋白质编码区,但也有相当一部分来自非编码区,如假基因和 lncRNA(图 5g,右)。
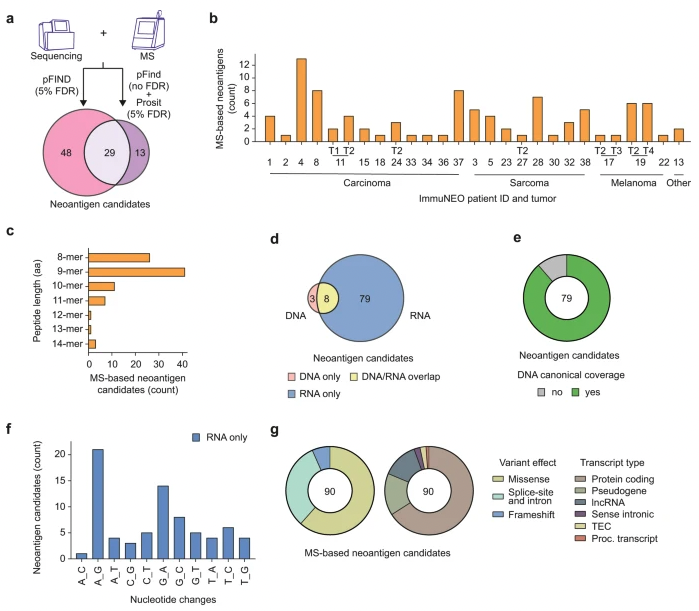
图5
综上所述,数据表明,基于MS的新抗原候选物识别对于大多数癌症患者来说是可行的,其中肿瘤 RNA 是检测源自变体的肽配体的重要来源。
5. RNA变异来源的新抗原具有免疫原性
为了评估已鉴定的新抗原候选物的免疫原性,研究通过体外试验评估了来自21名患者的78种新抗原候选物的 T 细胞反应,这些患者使用了自体或同种异体 HLA 匹配的外周血单核细胞 (PBMC) 或扩增的 TIL,通过 ELISpot 分析进行分析。
在 78 种检测的新抗原候选物中,有 21 种能够在自体 PBMC(图 6a,左)、扩增的 TIL(图 6a,右)或同种异体匹配的 PBMC(图 6b)培养环境中诱导 T 细胞反应(占所有测试的新抗原候选物的 27%)(图 6c)。大多数免疫原性新抗原是通过使用自体 PBMC 鉴定出来的,而只有三种免疫原性新抗原可以用扩增的 TIL 鉴定出来(图 6a)。这凸显了 已知的TIL 培养的困难,这可以通过扩增不足或扩增的 TIL 的 T 细胞表型失调和耗竭来解释,从而阻止了对所呈现的新抗原候选物的适当 T 细胞反应。尽管同种异体匹配的PBMC培养具有挑战性,特别是在供体选择方面,但研究测试了一小组新抗原候选物(n = 10),并可以确认四种在自体环境中具有免疫原性的新抗原的免疫原性,甚至还鉴定出一种额外的免疫原性新抗原(候选物 19A)(图 6b)。
所有21种免疫原性新抗原均从 RNA 来源中鉴定出来,其中20种仅从 RNA 变异中检测到,只有 一种从 RNA 和 DNA 变异中检测到(图 6d)。这支持了研究者的假设,即 RNA 改变机制可能与能够诱导患者 T 细胞反应的新抗原的形成有关。此外,产生免疫原性新抗原的变体的变异效应和转录本类型也与新抗原候选物的分布高度相似(图 6e)。
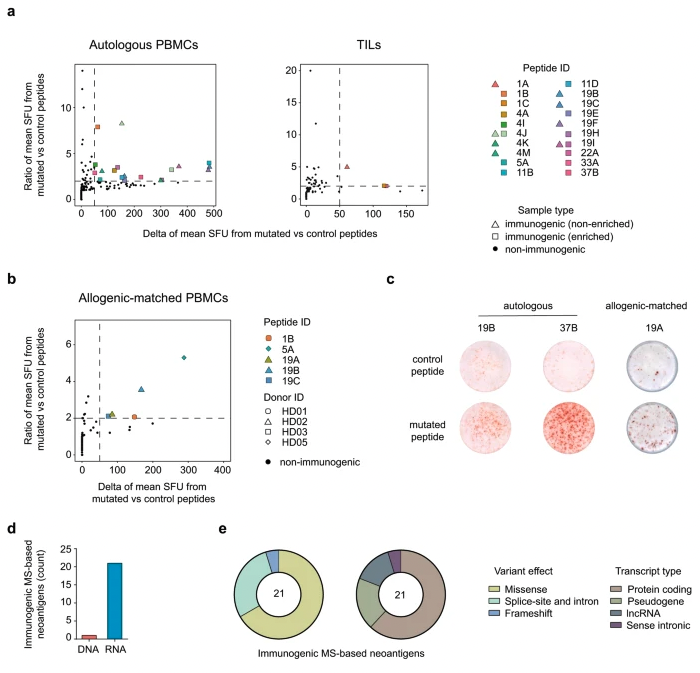
图6
总之,研究者利用利用肿瘤标本的 RNA 转录组学进行变异识别的蛋白质组学流程,在全癌症队列中四分之一的患者中鉴定出了独立于肿瘤实体的免疫原性新抗原。
6. 验证具有治疗潜力的新抗原候选肽
深入验证使得能够对非常有前景的新抗原候选物进行精细筛选。为了提高通过蛋白质基因组学管道检测潜在新抗原的可能性,研究者采用了较为宽松的初步筛选标准,这也是该领域的常见做法,因为这些靶标的出现率极低。然而,这可能增加发现假新抗原候选物的风险。因此,研究者将蛋白质基因组学分析流程作为假设生成工具,用于识别需要进一步验证的候选物。这里所应用的验证策略可能为临床转化中新抗原候选物的优先排序提供潜在的指导。
为了进一步确认谱图,研究者通过比较肿瘤中的新抗原候选物的内源性质谱(MS)谱图与其对应的合成肽的质谱谱图来进行肽序列验证。同时,使用Prosit预测的碎片离子强度进行了相同的比较,将归一化谱角(SA)达到0.7的肽为候选新抗原。经过88个测试肽的验证,研究者发现41个肽符合这些标准(图7)。有19个候选物接近SA的临界值,虽然仍有可能是真正的肽-谱匹配,但可能需要进一步确认。其他SA值较低的候选物可能不够安全用于治疗靶向,因为这些谱图可能被误认为是其他肽。未能验证的候选物很可能是由于研究者初步筛选标准较宽松。然而,尽管使用更严格的肽谱匹配标准(FDR低于1%)减少了SA值低于0.7的肽数量,研究者仍观察到许多SA值高于0.7、q值在1%到5%之间的肽(补充图10,红色矩形),如果研究者最初采用更严格的标准而非随后进行肽验证,这些肽本可能会被遗漏。此外,研究者将液相色谱(LC)实验的保留时间(RT)与预测的RT进行了比较,并将其作为肽验证的附加评分参数(图7和补充图11)。实验RT与预测RT相匹配的候选物占了一半(n = 45个候选物),而有一部分候选物的RT范围低于20分钟,在这一范围内基于所有测量的肽,预测算法的准确性较低(n = 17)。因此,RT偏差的候选物如果通过了SA临界值,可能不一定需要被排除。
由于RNA编辑是一种在健康中起重要作用的生理过程,研究者通过分析候选变异在正常组织中的情况,应用了进一步的新抗原确认步骤,使用公共数据库进行分析。研究者分析了来自Genotype-Tissue Expression (GTEx)项目的超过10,000个RNA-seq样本,涵盖30种不同组织,以检测90个新抗原变异的存在。在90个候选中,有38个在研究者的GTEx分析中完全未出现在正常组织的RNA-seq样本中,根据这一标准,它们可能是非常有趣的新抗原候选。其余52个候选物在正常组织中显示出高(n = 16,超过5%的样本中发现)、中(n = 6,在1%到5%的样本中发现)、低(n = 12,在0.1%到1%的样本中发现)、非常低(n = 7,低于0.1%的样本中发现)的流行率,或者被定义为不可用(N/A)(n = 9,在少于5%的正常组织样本中,变异位点有至少3个reads的表达)(图7)。正如预期,研究者未能在正常组织中检测到DNA来源的变异(图7)。
通过将这些肽验证和正常组织中的流行情况的质量控制整合到研究者的蛋白质基因组学新抗原鉴定流程中,研究者将新抗原候选分为20个非常有前景的候选(图7,顶部),12个需要进一步验证肽序列或在正常组织中的流行情况的潜在有前景候选(图7,中部),以及58个肽验证不足或在正常组织中常见的候选(图7,底部)。大多数32个验证候选由pFind和Prosit共同鉴定,并且主要是九肽(补充图14a-c)。重要的是,大多数验证的新抗原候选来自缺乏体细胞突变的RNA变异,可能是RNA编辑事件(补充图14d-f)。在验证的候选物中,蛋白质编码变异是最常见的转录类型(补充图14g)。当使用NetMHC 4.0和MHCFlurry对研究者鉴定的新抗原候选进行结合预测时(补充数据4),90%的非常有前景候选(20个中的18个),58%的潜在有前景候选(12个中的7个),以及51%的未验证候选(59个中的30个)至少在一种算法中被预测为结合体(百分位排名<2%或预测结合亲和力<500 nM)。这一验证候选中结合体的富集支持了研究者的肽验证方法表现良好。在32个验证候选中,8个引发了CD8+ T细胞的免疫反应。
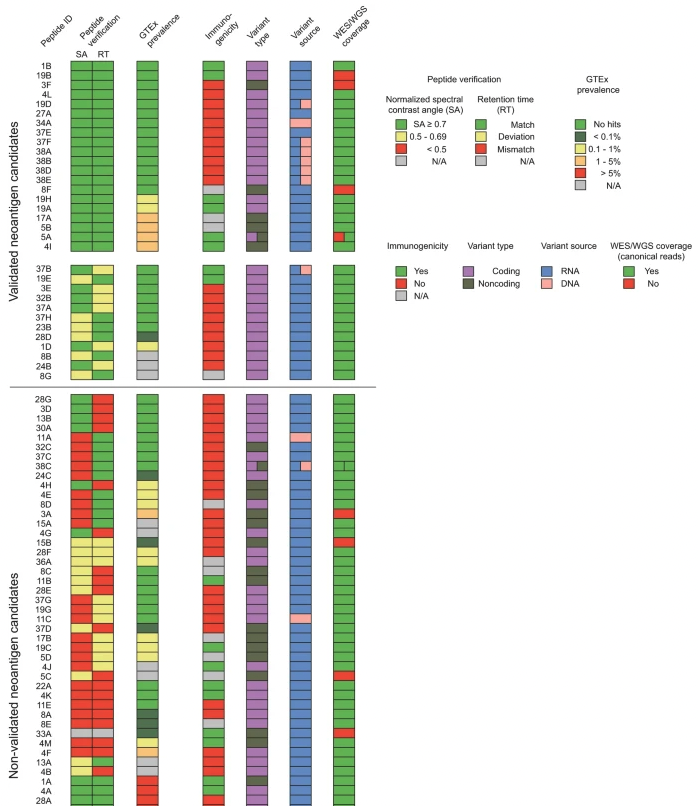
图7
总之,研究者在肿瘤新抗原识别流程中加入了全面的验证分析,为免疫疗法靶标的选择提供了重要参考。这些免疫肽与 T 细胞浸润和潜在的 T 细胞衰竭表型相关。

汇报人:程丹妮
导师:赵宇
审核:饶郁芳、邱轲、任建君
