


精读分享│:冷冻休克的肿瘤细胞通过合成致死性递送CRISPR-Cas9以实现肺癌消退
英文题目:Cryo-shockedtumorcellsdeliverCRISPR-Cas9forlungcancerregressionbysyntheticlethality
中文题目:冷冻休克的肿瘤细胞通过合成致死性递送CRISPR-Cas9以实现肺癌消退
期刊:ScienceAdvance(IF=12.5)

Abstract
Although CRISPR-mediated genome editing holds promise for cancer therapy, inadequate tumor targeting and potential off-target side effects hamper its outcomes. In this study, we present a strategy using cryo-shocked lung tumor cells as a CRISPR-Cas9 delivery system for cyclin-dependent kinase 4 (CDK4) gene editing, which initiates synthetic lethality in KRAS-mutant non-small cell lung cancer (NSCLC). By rapid liquid nitrogen shocking, we effectively eliminate the pathogenicity of tumor cells while preserving their structure and surface receptor activity. This delivery system enables the loaded CRISPR-Cas9 to efficiently target the lung through capture in pulmonary capillaries and interactions with endothelial cells. In a NSCLC-bearing mouse model, drug accumulation in the lung is increased nearly fourfold, and intratumoral CDK4 expression is substantially down-regulated compared to CRISPR-Cas9 administration via lipofectamine nanoparticles. Furthermore, CRISPR-Cas9 editing-mediated CDK4 ablation triggers synthetic lethality in KRAS-mutant NSCLC and prolongs the survival of mice.
摘要
CRISPR介导的基因组编辑为癌症治疗带来了广阔前景,但肿瘤靶向性不足及潜在脱靶效应仍制约其临床治疗和转化效果。本研究提出一种新型递送策略:以快速冷冻冲击处理的肺部肿瘤细胞为载体,递送CRISPR-Cas9系统,靶向编辑细胞周期蛋白依赖性激酶4(CDK4)基因,从而在KRAS突变的非小细胞肺癌(NSCLC)中引发合成致死效应。通过快速的液氮冲击,肺部肿瘤细胞的致病性被有效清除,同时其细胞结构完整性及表面受体活性得以保留。该递送系统通过在肺毛细血管中的捕获以及与内皮细胞的相互作用,使负载的CRISPR-Cas9能高效靶向富集至肺部。在NSCLC荷瘤的小鼠模型中,该策略使肺部药物累积量较传统递送方式提高了近四倍;且与Lipofectamine纳米粒子介导的CRISPR-Cas9递送相比,肿瘤组织中CDK4表达水平显著下调。此外,CRISPR-Cas9编辑介导的CDK4敲除在KRAS突变NSCLC中特异性诱发合成致死反应,延长了小鼠的生存期。
研究背景:
对于许多晚期或转移性NSCLC患者而言,由于该病缺乏特定治疗靶点,而目前临床治疗手段有限,患者的中位总生存期极低。CRISPR-Cas9能够通过永久性地破坏肿瘤生存关键基因,因此在诊断和治疗各种肿瘤、病毒感染和遗传性疾病展现出巨大的潜力,该技术理论上无需重复给药。
然而,CRISPR-Cas9的临床应用受多重瓶颈制约:一方面,CRISPR-Cas9系统在血液中易发生降解或变性,导致传递效率低;另一方面,目前主要的递送载体,包括病毒载体和非病毒载体,普遍缺乏组织靶向性和细胞选择性,成为限制其体内应用的关键障碍。
合成致死(syntheticlethality)机制为解决上述困境提供了重要理论基础,其被定义为两个基因同时失活导致细胞死亡,而单独敲除其中任一基因对细胞生存无显著影响。一般来说,肿瘤细胞携带特定癌基因突变,这使其对维持细胞稳态的某些基因产生依赖性。原则上,这些携带特定突变的肿瘤细胞可以通过药物靶向抑制与其具有合成致死相互作用的另一个基因而被选择性地杀死,而由于缺乏特定的遗传变异,正常细胞可以免受药物的影响。因此,合成致死为靶向不可治疗的癌基因提供了有前途的治疗策略,同时减轻了对正常组织和细胞的损害。
研究方法和思路:
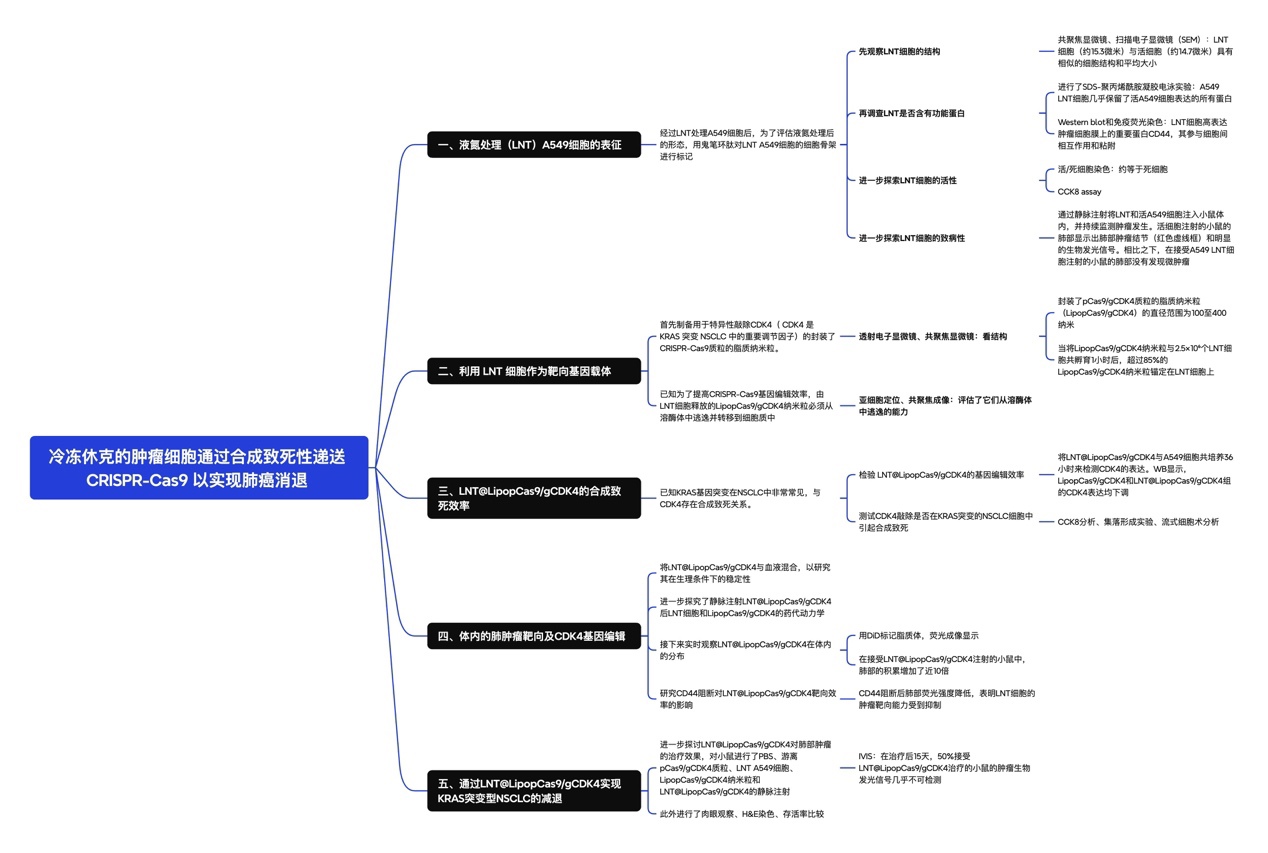
研究方法:
一、液氮处理(LNT)A549细胞的制备、表征及生物安全性验证
A549细胞是一种典型的KRAS突变型人非小细胞肺癌细胞(NSCLC)细胞。首先经过液氮快速冷冻灭活(LNT)处理该细胞,在消除其致病性的同时,保留细胞结构及表面功能蛋白活性。
LNT-A549细胞制备:
收集对数生长期A549细胞,悬浮在冻存液中,置于液氮中静置冷冻12小时。使用前,将冻存细胞在冰浴中快速解冻,37°C条件下复温后,以500g离心离心3min,弃上清,用磷酸盐缓冲液(PBS)洗涤2次,重悬备用。
LNT-A549细胞表征:
(1)细胞结构观察:
共聚焦显微镜:取1×10⁶个LNT-A549细胞,用4%多聚甲醛固定10分钟,加入含6.6μMAF488标记鬼笔环肽的PBS溶液500μl,室温孵育20分钟,500g离心3分钟收集细胞;用含1μg/mlDAPI的PBS溶液室温孵育10分钟,对细胞核进行染色;PBS洗涤后重悬,采用ZeissLSM800共聚焦显微镜观察细胞骨架及细胞核形态。结果显示,LNT-A549细胞平均直径约15.3μm,与活A549细胞(平均直径14.7μm)的细胞形态、骨架结构及细胞核形态无显著差异(图1B、C)
扫描电子显微镜(SEM):LNT-A549细胞经2.5%的戊二醛固定30分钟后,依次使用30%、50%、70%、85%和90%梯度乙醇脱水,每级处理10分钟,再用100%乙醇洗涤2次,每次30分钟。将脱水后的LNT-A549细胞滴在硅片上,通过NovaNano450扫描电子显微镜进一步分析。SEM图像显示,LNT-A549细胞呈球形结构,表面粗糙度与活A549细胞相似,证实液氮处理未破坏细胞结构完整性,保持了活细胞的结构(图1D)。
(2)细胞活性分析:
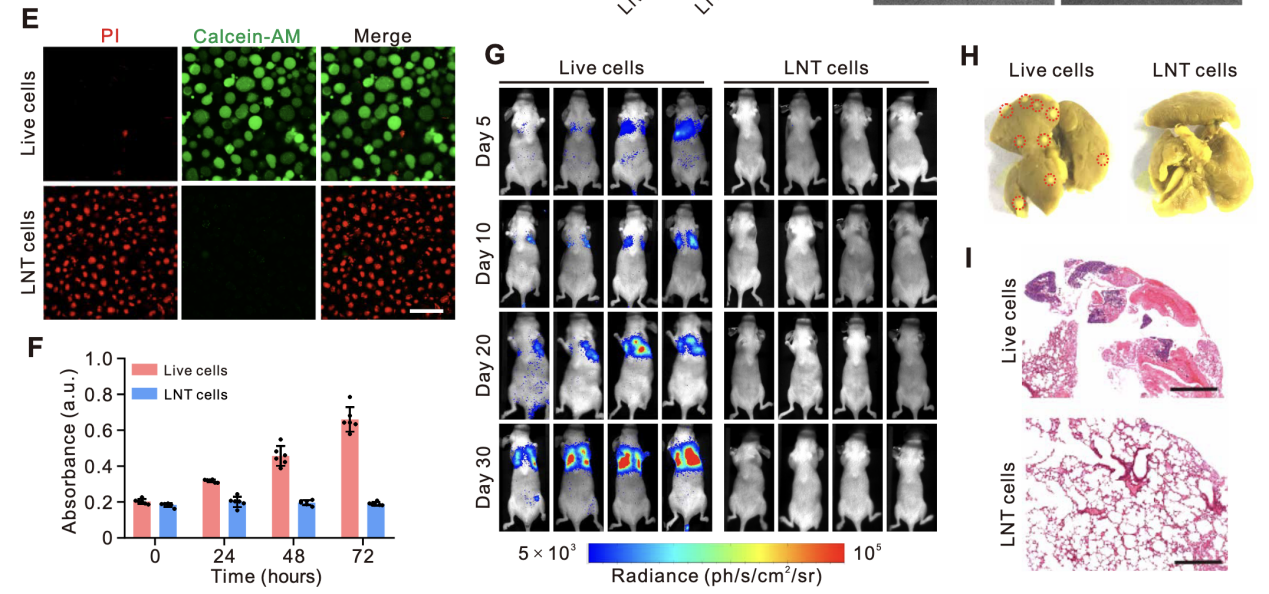
活/死细胞染色:采用钙黄绿素乙酰氧甲酯(calcein-AM)/碘化丙啶(PI)双染法,将LNT-A549细胞与活A549细胞分别接种于共聚焦培养皿,加入适量双染试剂,37℃孵育30分钟,激光扫描共聚焦显微镜下观察荧光信号(活细胞呈绿色荧光,死细胞呈红色荧光)。calcein-AM/PI双染结果显示,LNT-A549细胞均呈现红色荧光,无绿色荧光信号,表明细胞已完全灭活(图1E)。
CCK-8活性评估:将LNT-A549细胞与活A549细胞分别以相同密度接种于96孔板,常规培养0、1、2、3天;每孔加入10μlCCK-8试剂,37℃孵育2小时,使用酶标仪检测450nm处吸光度值(OD₄₅₀),评估细胞增殖活性。CCK-8实验显示,活A549细胞培养3天后OD₄₅₀值较初始水平升高了约3倍,而LNT-A549细胞在3天培养期内OD₄₅₀值无明显变化,进一步证实LNT处理可彻底消除细胞增殖活性(图1F)。
(3)细胞功能蛋白验证:
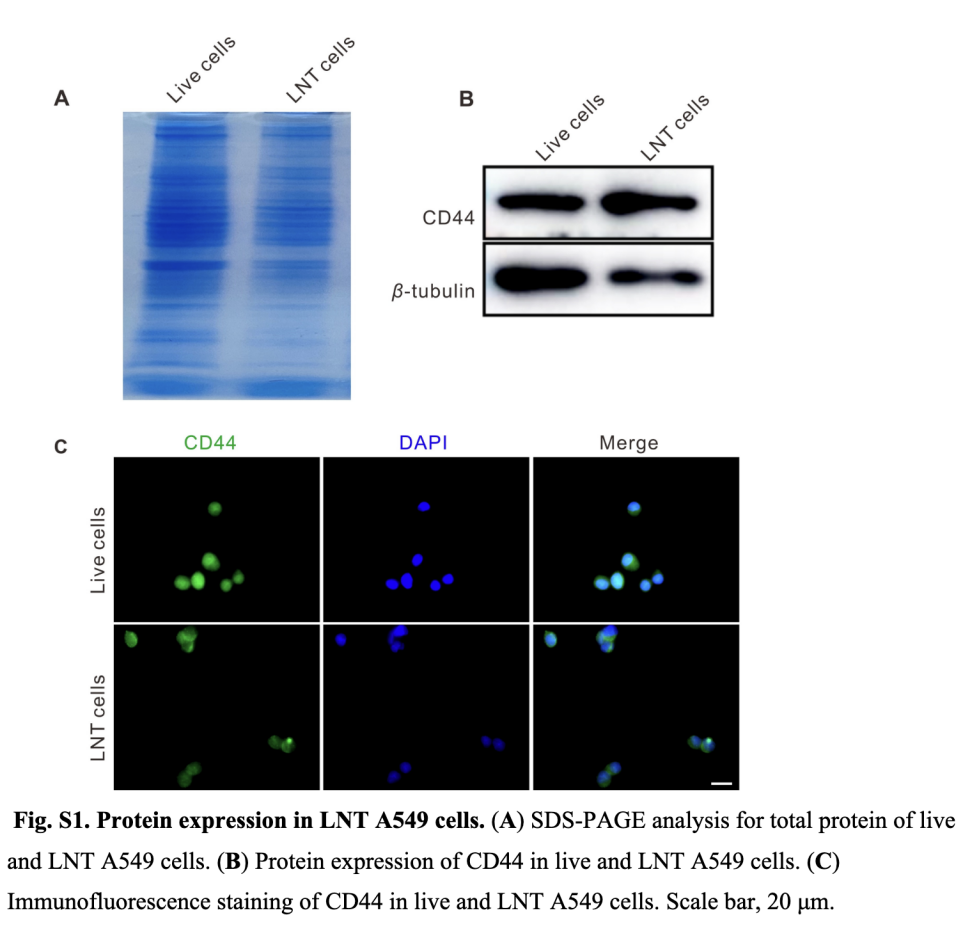
SDS-聚丙烯酰胺凝胶电泳(SDS-PAGE):分别提取LNT-A549细胞与活A549细胞的总蛋白,经蛋白定量后,取等量蛋白进行SDS-PAGE电泳,考马斯亮蓝染色后观察蛋白条带分布。SDS-PAGE电泳结果显示,LNT-A549细胞的蛋白条带图谱与活A549细胞高度一致,其几乎保留了活细胞所有表达的蛋白(图S1A)。
Western blot检测:提取细胞总蛋白,经SDS-PAGE电泳后转印至PVDF膜,封闭后加入CD44一抗及相应二抗孵育,ECL化学发光法检测CD44蛋白表达水平。Western blot及免疫荧光染色结果显示,LNT-A549细胞中CD44蛋白仍保持高表达水平,其参与细胞间相互作用和粘附(图S1B、C),证实液氮处理未影响细胞表面关键功能蛋白的表达及定位。
免疫荧光染色:LNT-A549细胞固定后,加入CD44一抗孵育过夜,荧光标记二抗室温孵育1小时,DAPI染核后,共聚焦显微镜观察CD44蛋白的定位及表达。
体内生物安全性验证
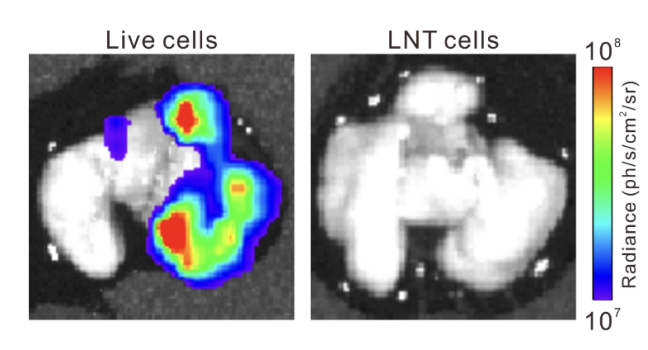
将LNT-A549细胞与活A549细胞分别通过尾静脉注射入裸鼠体内,持续监测小鼠生存状态及肺部肿瘤形成情况,解剖分离肺部组织,观察肿瘤结节形成,并通过生物发光成像技术检测肿瘤信号强度。体内注射实验显示,活A549细胞注射组小鼠肺部出现明显肿瘤结节,且检测到强生物发光信号;而LNT-A549细胞注射组小鼠肺部未发现微肿瘤,生物发光信号阴性(图1G-I、图S2),表明冷冻冲击的A549细胞在体外和体内都具有生物安全性。
二、EZH2调节乳腺癌骨转移的恶性循环
图a展示了在TGFβ刺激下,将乳腺癌细胞与破骨前细胞和成骨细胞三重共培养,模拟乳腺癌骨转移微环境的恶性循环。从图b显示,敲除了EZH2的肿瘤细胞生长被抑制。同时,图c的结果揭示了,敲除EZH2会使得成熟破骨细胞变少。为了检测EZH2甲基转移酶在乳腺癌骨转移恶性循环中的作用,图d中敲除EZH2的肿瘤细胞再用H689A(EZH2甲基转移酶死亡突变体)处理之后,与野生
EZH2在TGFβ刺激下增加pS465/467-Smad2和pY397-FAK水平
LNT细胞作为靶向基因载体介导CRISPR-Cas9递送
CDK4是KRAS突变NSCLC细胞的重要调节因子,本研究构建了封装CRISPR-Cas9质粒以特异性敲除CDK4的脂质纳米粒。
LipopCas9/gCDK4纳米粒制备:
采用薄膜分散法制备脂质纳米粒,将阳离子脂质、辅助脂质按一定摩尔比溶解于氯仿中,旋转蒸发去除有机溶剂形成均匀脂质膜;加入含pCas9/gCDK4质粒的无菌水,振荡水化后通过挤出器挤压,获得均一分散的LipopCas9/gCDK4纳米粒。
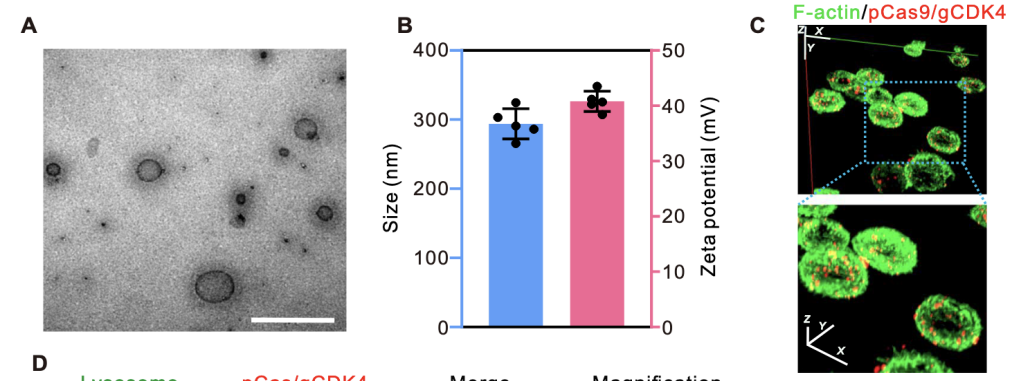
透射电子显微镜(TEM):取适量LipopCas9/gCDK4纳米粒悬液滴加至铜网上,磷钨酸负染后,通过透射电子显微镜(TecnaiG2F20)观察纳米粒形态并测量粒径范围。TEM图像显示,LipopCas9/gCDK4纳米粒呈类球形均匀分布,粒径范围为100~400nm(图2A)。
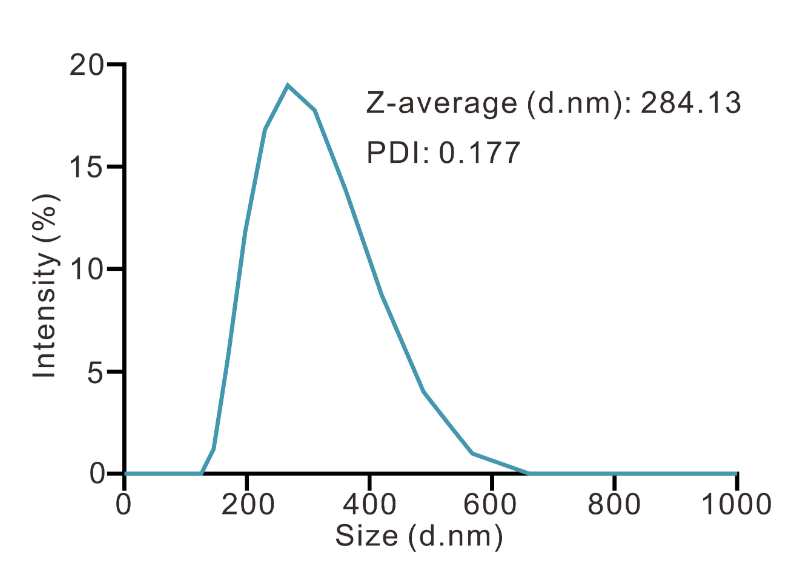
动态光散射(DLS):采用马尔文激光粒度仪测定LipopCas9/gCDK4纳米粒的粒径分布、平均粒径、zeta电位及聚合物分散指数(PDI),每个样本检测3次,取平均值。DLS检测结果显示,其平均粒径为294nm,zeta电位为40.8mV,聚合物分散指数(PDI)为0.177,表明纳米粒具有良好的分散均一性(图2B、图S3)。
LNT@LipopCas9/gCDK4递送系统构建:
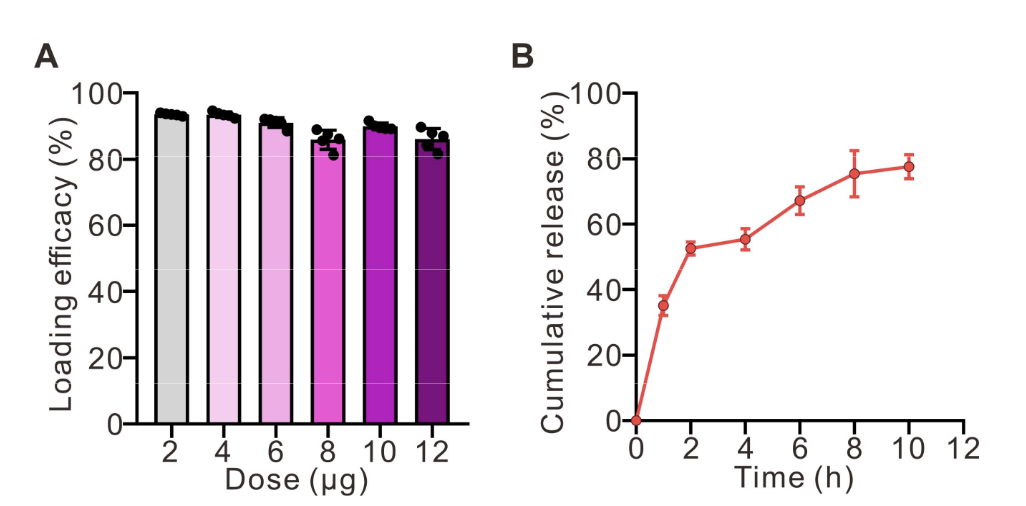
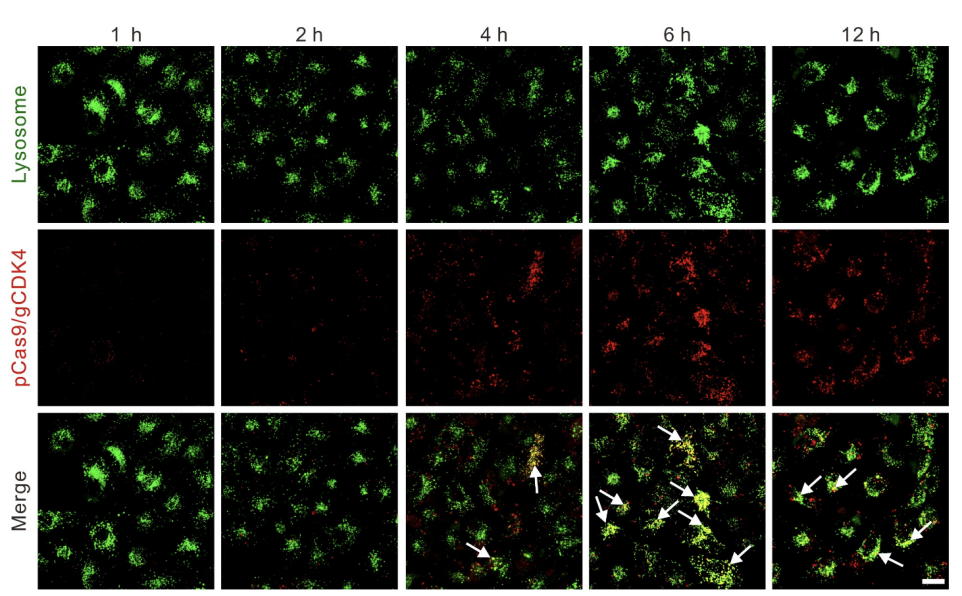
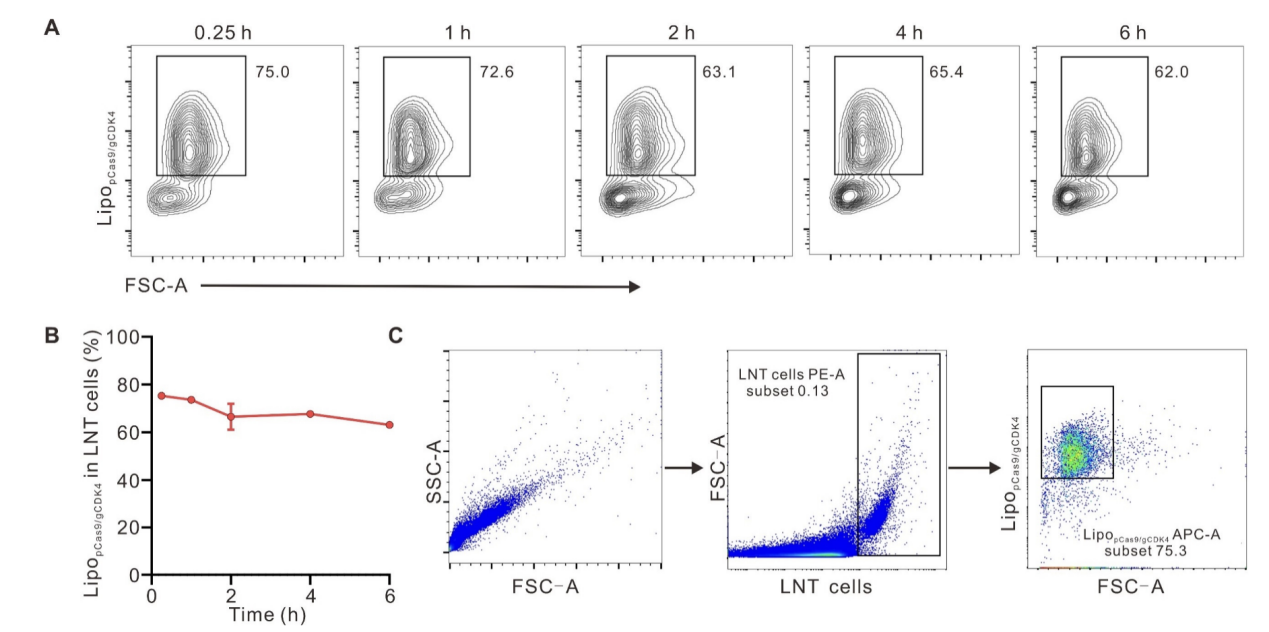
首先进行质粒标记,用iFluor647荧光染料对pCas9/gCDK4质粒进行共价标记,透析纯化后备用。随后进行纳米粒细胞锚定,将2.5×10⁶个LNT-A549细胞与LipopCas9/gCDK4纳米粒悬液按优化比例混合,37℃孵育1小时,通过静电相互作用实现纳米粒在细胞表面的锚定;孵育结束后,PBS洗涤3次以去除未结合的游离纳米粒。共聚焦显微镜成像显示,iFluor647标记的LipopCas9/gCDK4纳米粒在LNT-A549细胞表面呈现清晰的红色荧光信号,证实纳米粒通过静电相互作用成功锚定在细胞表面(图2C);荧光定量检测显示,2.5×10⁶个LNT-A549细胞与纳米粒共孵育1小时后,锚定效率高达85%(图S4A),且该复合物在体外模拟生理环境中表现出持续释放特性,10小时内pCas9/gCDK4的累积释放率约为78%(图S4B),为后续肿瘤部位的基因编辑提供了持续的药物供给。
溶酶体逃逸能力评估:
将LNT@LipopCas9/gCDK4复合物与A549细胞共培养,分别在2、6、12小时加入溶酶体特异性探针(LysoTrackerGreen)孵育30分钟,DAPI染核后,通过ZeissLSM800共聚焦显微镜观察iFluor647标记的质粒(红色荧光)与溶酶体(绿色荧光)的共定位情况,计算共定位系数。亚细胞定位结果显示,共培养2小时时,细胞内红色荧光信号微弱,推测此时纳米粒尚未从LNT-A549细胞表面脱落(图S5);6小时时,红色荧光与溶酶体绿色荧光的共定位系数较高,表明部分LipopCas9/gCDK4纳米粒被A549细胞吞噬并包裹于溶酶体中;12小时时,两者共定位系数显著降低,红色荧光在细胞质中呈弥散分布,证实相当一部分纳米粒成功实现溶酶体逃逸(图2D)。
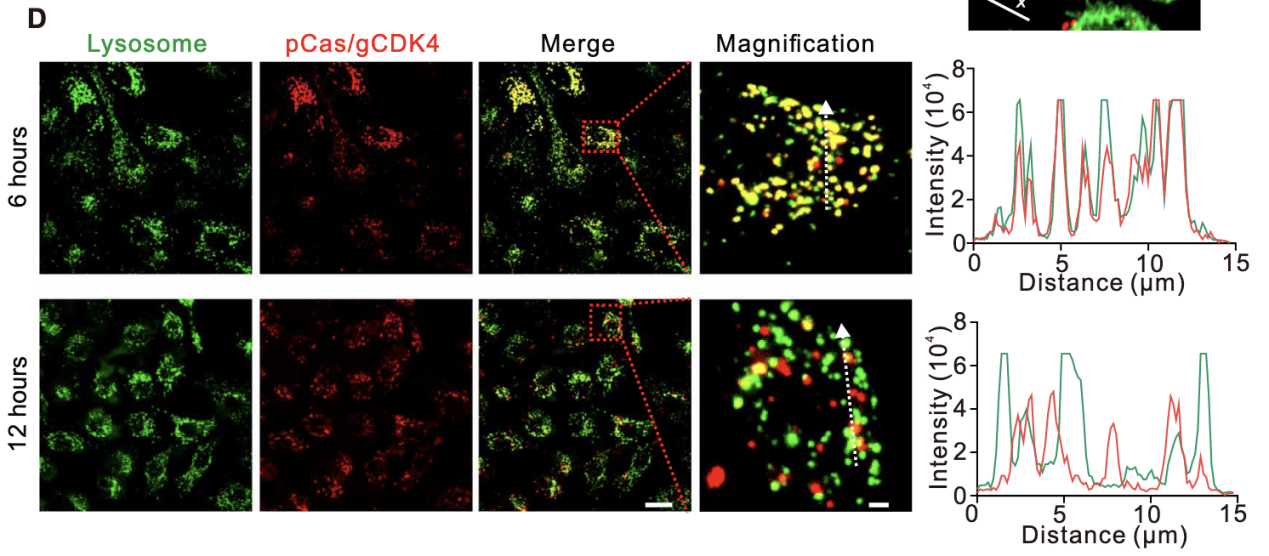
LNT@LipopCas9/gCDK4的合成致死效率
KRAS基因突变是NSCLC中最常见的驱动突变之一,已证实KRAS突变与细胞周期蛋白依赖性激酶4(CDK4)之间存在明确的合成致死关系——KRAS突变型肿瘤细胞的存活高度依赖CDK4功能,而CDK4敲除对KRAS野生型细胞无显著影响。本研究通过体外实验验LNT@LipopCas9/gCDK4递送系统的CDK4基因编辑效率,并明确其在KRAS突变NSCLC中的合成致死效应。
LNT@LipopCas9/gCDK4基因编辑效率:
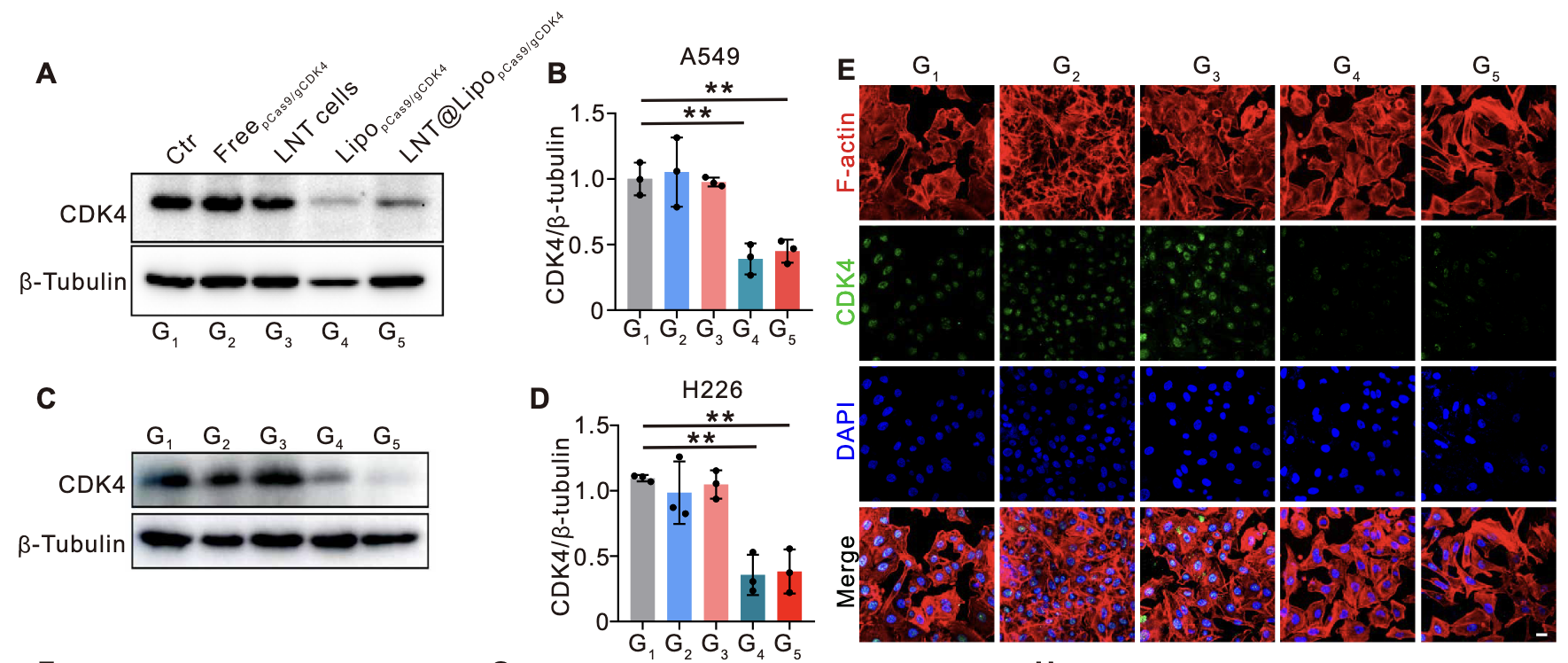
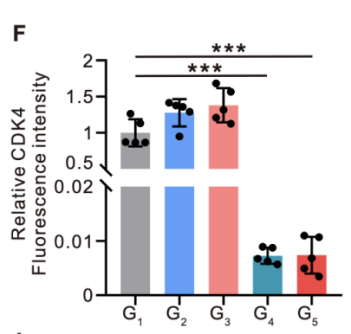
Western blot检测:将LNT@LipopCas9/gCDK4复合物与KRAS突变型NSCLC细胞系(A549)共培养36小时后,提取细胞总蛋白进行Western blot分析。结果显示,LipopCas9/gCDK4纳米粒组与LNT@LipopCas9/gCDK4组的CDK4蛋白表达水平均显著下调;而单纯LNT细胞处理组及游离pCas9/gCDK4质粒组的CDK4表达无明显变化,表明仅功能性CRISPR-Cas9递送系统可实现有效的CDK4敲除(图3A、B)。同时为明确编辑特异性,在KRAS野生型人肺鳞癌细胞系(H226)中进行平行实验。结果显示,LNT@LipopCas9/gCDK4处理后H226细胞的CDK4表达未出现显著下调(图3C、D),提示该递送系统的编辑效应不影响KRAS野生型细胞的CDK4表达,为后续合成致死的特异性提供了基础。
免疫荧光验证:对LNT@LipopCas9/gCDK4处理后的A549细胞进行CDK4免疫荧光染色,共聚焦显微镜观察显示,处理组细胞的CDK4特异性荧光强度显著弱于对照组,进一步从细胞水平证实了CDK4敲除的有效性(图3E、F)。
合成致死效应评估:
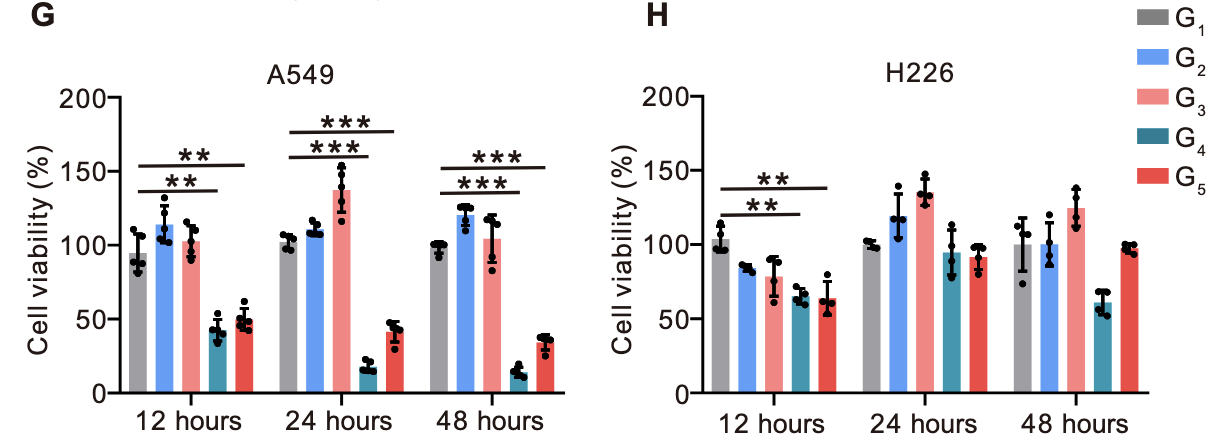
细胞活性检测(CCK-8实验):将LNT@LipopCas9/gCDK4与A549细胞共培养后,使用CCK-8法检测细胞活性。结果显示,处理24小时后,LipopCas9/gCDK4组与LNT@LipopCas9/gCDK4组的A549细胞活性均下降约50%;延长处理时间至48小时,两组死亡细胞比例均超过90%,表明CDK4敲除可有效抑制KRAS突变型NSCLC细胞存活(图3G)。而在H226细胞中,LNT@LipopCas9/gCDK4处理后细胞活性仅出现轻微下降,证实合成致死效应具有KRAS突变依赖性(图3H)。
细胞增殖抑制(集落形成实验):LNT@LipopCas9/gCDK4处理后,A549细胞的集落形成数量显著减少,集落体积明显缩小;而H226细胞的集落形成能力未受明显影响(图3I),进一步验证了CDK4敲除对KRAS突变型肿瘤细胞增殖的特异性抑制作用。
细胞周期分析(流式细胞术):流式细胞术检测细胞周期分布显示,A549细胞经LipopCas9/gCDK4或LNT@LipopCas9/gCDK4处理24小时后,G1/S期细胞比例显著升高,细胞周期阻滞明显加剧(图3J)。
体内的肺肿瘤靶向及CDK4基因编辑效率验证
肿瘤细胞在循环系统中具有易滞留于肺毛细血管的生理特性,结合LNT-A549细胞高表达CD44粘附蛋白的特性,本研究通过体内实验验证LNT@LipopCas9/gCDK4递送系统的血液稳定性、体内分布特征、肺肿瘤靶向机制及CDK4基因编辑效率,为其体内治疗应用提供依据。
血液稳定性与药代动力学分析:
血液稳定性:将LNT@LipopCas9/gCDK4复合物与新鲜小鼠全血混合,以研究其在生理条件下的稳定性。结果显示,混合孵育后大多数LipopCas9/gCDK4纳米粒仍稳定锚定于LNT细胞表面,未出现明显脱落(图S6),表明LNT细胞与LipopCas9/gCDK4之间存在稳定的相互作用。
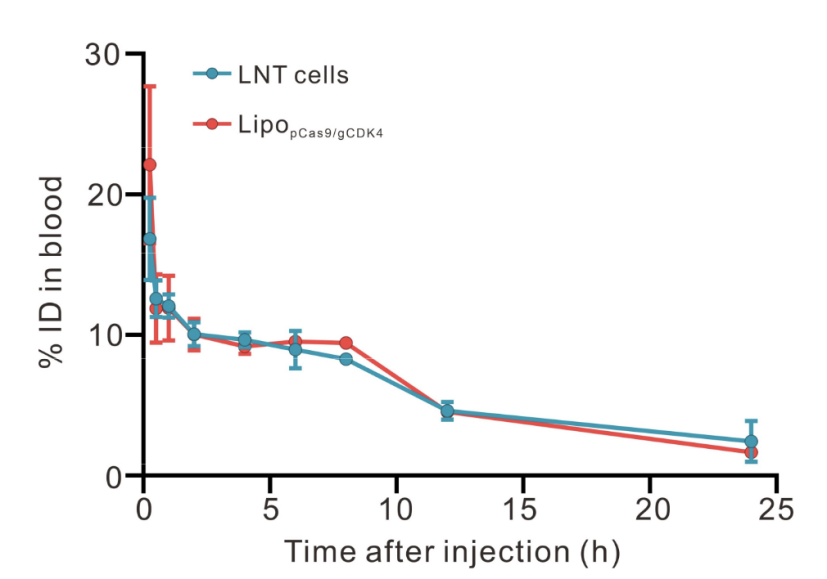
药代动力学特征:通过尾静脉注射LNT@LipopCas9/gCDK4复合物,动态监测LNT细胞(荧光标记)与LipopCas9/gCDK4纳米粒(荧光标记)的体内清除曲线。结果显示,两组药代动力学曲线趋势高度一致,表明LNT细胞与锚定的纳米粒在体内的循环和清除都是同步进行的,进一步证实两者间相互作用的稳定性(图S7)。
2.体内肺肿瘤靶向性评估:
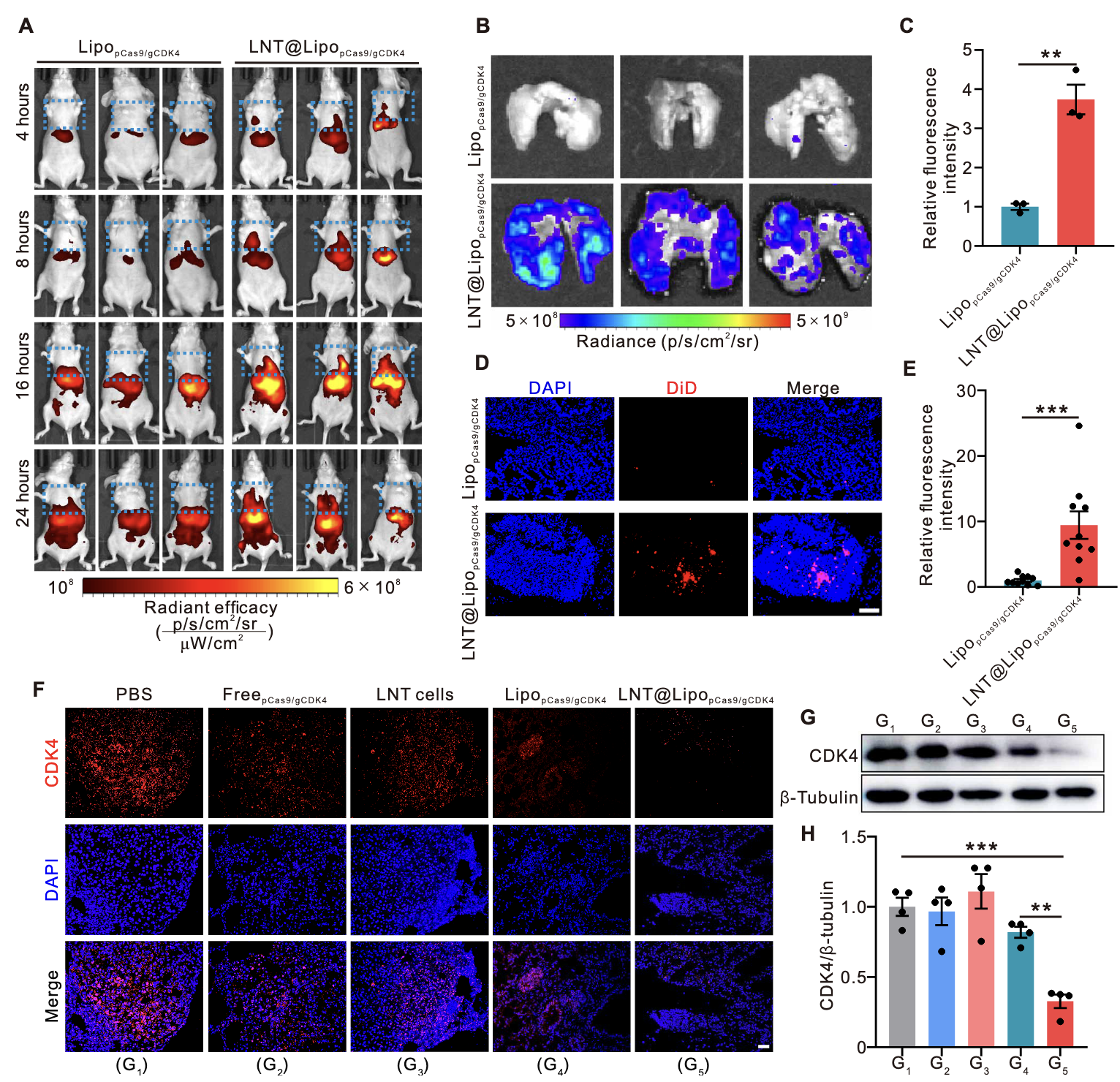
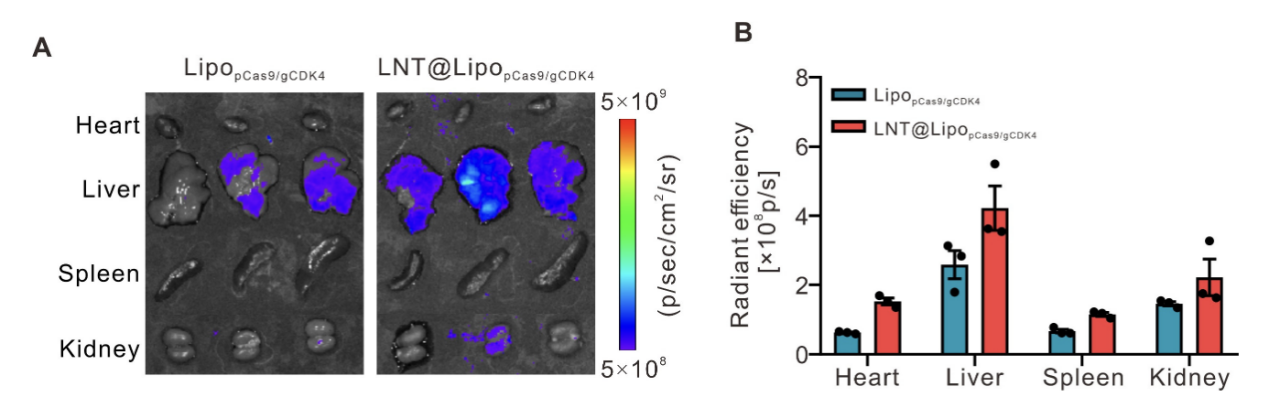
体内分布实时成像:采用DiD荧光染料标记LipopCas9/gCDK4纳米粒,通过活体荧光成像系统(IVIS)实时观察小鼠静脉注射后的体内分布。结果显示,注射后16小时内,LNT@LipopCas9/gCDK4组小鼠肺部荧光信号持续强于单纯LipopCas9/gCDK4组(图4A);离体肺部组织成像显示,注射6小时后,LNT@LipopCas9/gCDK4组肺部荧光信号强度约为LipopCas9/gCDK4组的4倍(图4B、C);病理切片荧光染色定量分析显示,LNT@LipopCas9/gCDK4组肺部药物累积量较LipopCas9/gCDK4组提升近10倍(图4D、E)。此外,该递送系统在肝脏、脾脏等其他主要器官的荧光信号虽略有升高,但远低于肺部富集水平(图S8),证实其具有显著的肺靶向特异性。
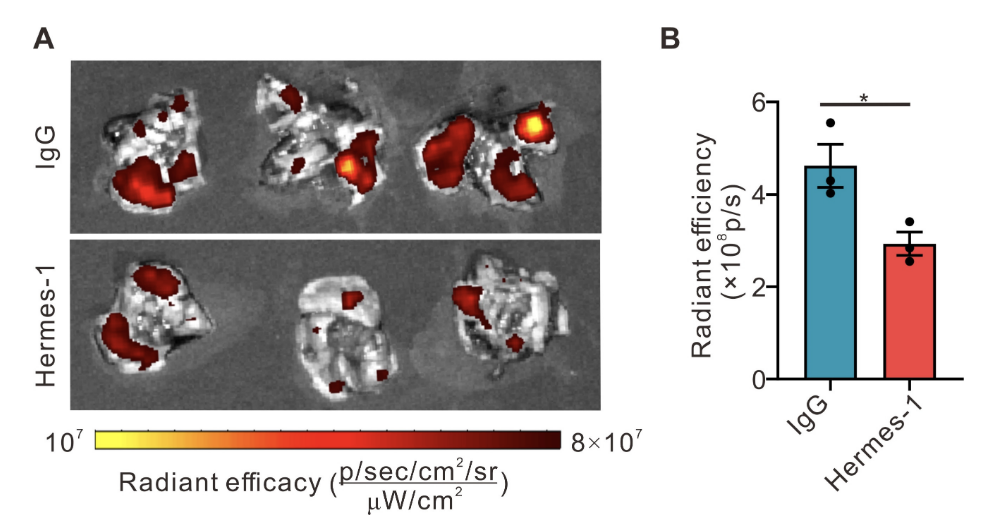
CD44依赖性的靶向机制验证:为明确CD44蛋白在靶向过程中的作用,采用CD44阻断剂Hermes-1预处理LNT细胞后,再构建LNT@LipopCas9/gCDK4复合物并注射入NSCLC荷瘤小鼠体内。IVIS成像结果显示,CD44阻断后,小鼠肺部荧光信号强度显著降低(图S9),表明LNT细胞表面CD44蛋白通过介导细胞间粘附及与肺毛细血管内皮细胞的相互作用,增强了递送系统的肺肿瘤靶向能力,证实该靶向过程具有CD44依赖性。
体内CDK4基因编辑效率
取各组小鼠肺部肿瘤组织,通过免疫荧光染色及Westernblot(WB)检测CDK4蛋白表达水平。结果显示,与单纯LipopCas9/gCDK4组、LNT细胞组及安慰剂组相比,LNT@LipopCas9/gCDK4处理组的肿瘤组织中,CD44特异性荧光强度显著减弱(图4F、G),WB检测的CDK4蛋白条带灰度值显著降低(图4H),证实LNT@LipopCas9/gCDK4递送系统可在体内有效富集于肺肿瘤部位,并成功介导CDK4基因编辑,显著下调肿瘤组织中CDK4的表达。
五、LNT@LipopCas9/gCDK4介导的KRAS突变型NSCLC的肿瘤减退
1.体内肿瘤消退效果:
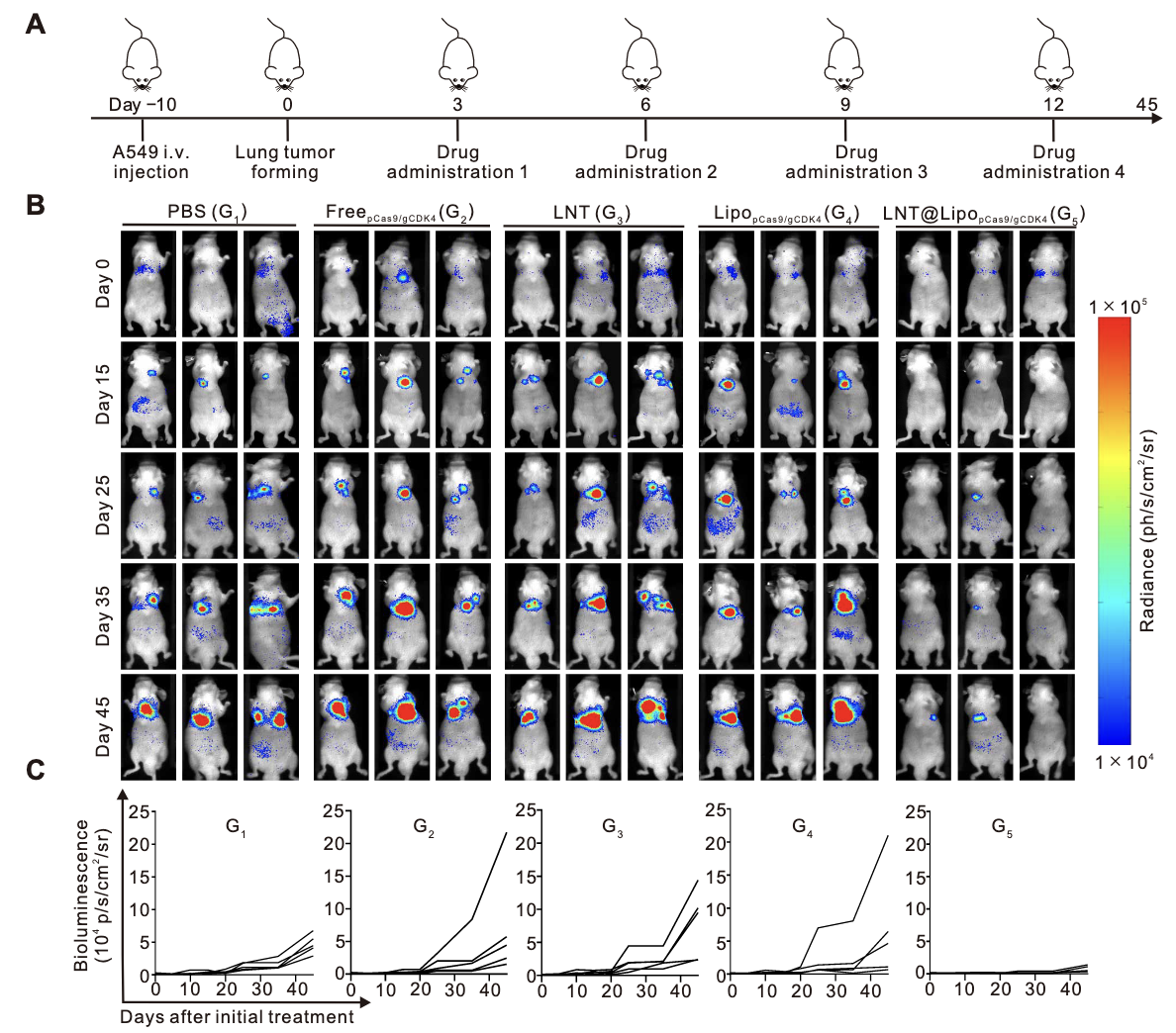
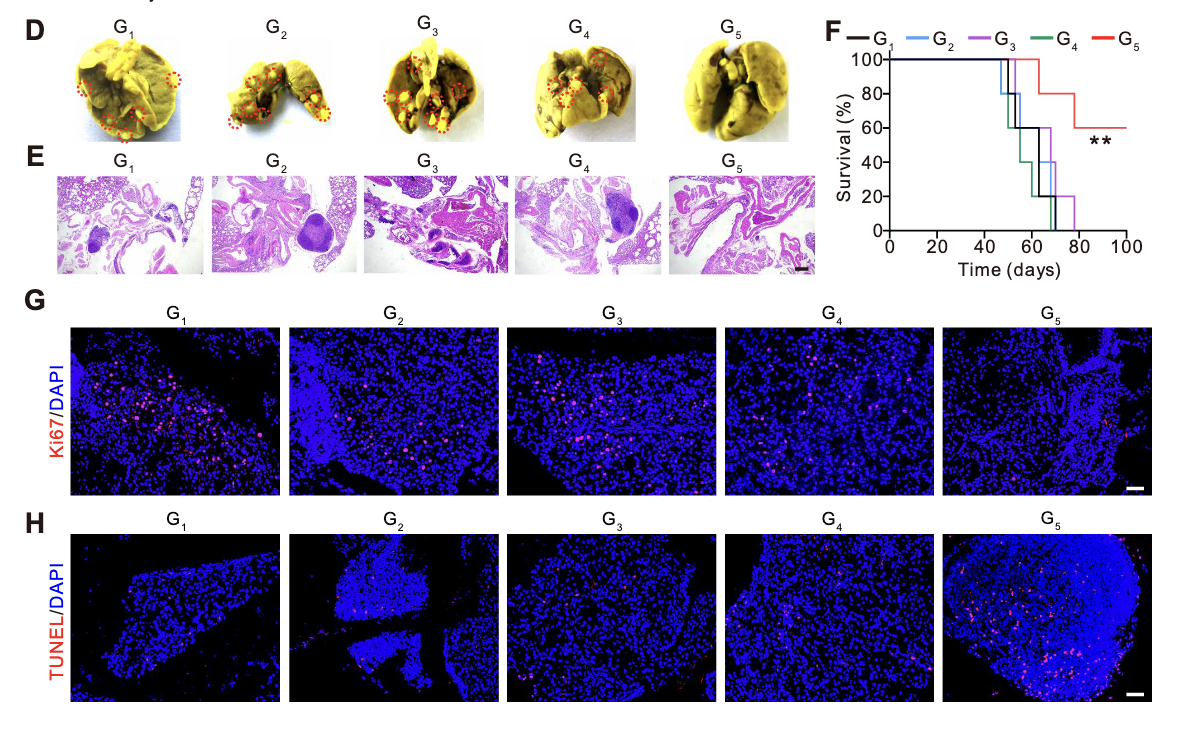
肿瘤生长动力学监测:采用活体生物发光成像系统(IVIS)实时追踪A549细胞来源肿瘤的生物发光信号,评估肿瘤生长状态。结果显示,LNT@LipopCas9/gCDK4组小鼠的肿瘤生物发光信号呈持续下降趋势,治疗后15天,50%的小鼠肿瘤信号几乎降至检测下限,表现出显著的肿瘤消退效应;而PBS组、游离质粒组、LNT细胞组及单纯纳米粒组的肿瘤生物发光信号持续升高,肿瘤呈快速进展态势(图5B、C),证实LNT@LipopCas9/gCDK4的靶向递送与合成致死协同作用可有效抑制肿瘤生长。
肿瘤结节计数与病理验证:治疗终点(肿瘤进展至伦理终点或治疗后60天)解剖小鼠肺部,肉眼观察及计数显示,LNT@LipopCas9/gCDK4组的肺部肿瘤结节数量显著少于其他各组(图5D);HE染色结果进一步证实,该组肿瘤组织体积缩小、细胞密度降低,而对照组肿瘤组织呈典型恶性增殖特征(图5E),从病理层面验证了治疗效果。
生存获益评估
小鼠存活曲线分析显示,LNT@LipopCas9/gCDK4组的生存获益显著优于其他各组:60%的小鼠存活时间达到100天,且未出现明显肿瘤复发迹象;而PBS对照组小鼠全部在70天内死亡,游离质粒组、LNT细胞组及单纯纳米粒组的中位生存期虽略有延长,但与LNT@LipopCas9/gCDK4组相比存在统计学差异(图5F),表明该治疗策略可显著延长KRAS突变型NSCLC荷瘤小鼠的生存期。
治疗机制体内验证
肿瘤增殖抑制:Ki67免疫荧光染色(细胞增殖标志物)结果显示,LNT@LipopCas9/gCDK4组肿瘤组织中Ki67阳性细胞比例显著降低(图5G),证实CDK4敲除介导的合成致死效应可有效抑制肿瘤细胞增殖。
肿瘤细胞凋亡诱导:末端脱氧核苷酸转移酶介导的脱氧尿苷三磷酸缺口末端标记(TUNEL)染色显示,LNT@LipopCas9/gCDK4组肿瘤组织中的凋亡细胞数量显著增加(图5H),表明该递送系统通过CDK4敲除不仅抑制肿瘤增殖,还可诱导肿瘤细胞凋亡,从而发挥双重抗肿瘤作用。
治疗安全性评估
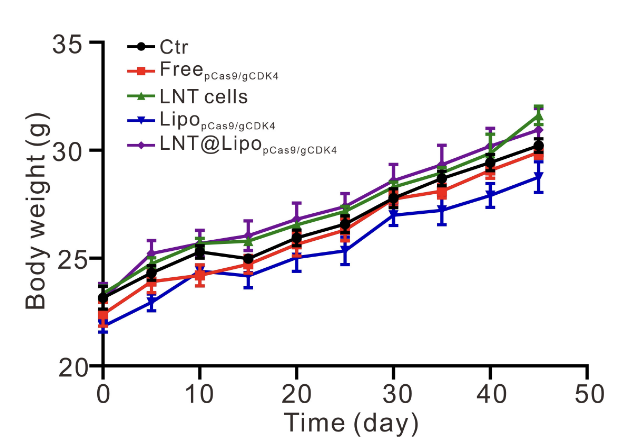
治疗期间每周监测小鼠体重变化,结果显示,各组小鼠体重均无显著波动(图S10),未出现体重下降、精神萎靡、食欲减退等不良反应,证实LNT@LipopCas9/gCDK4治疗策略具有良好的体内生物安全性,未对小鼠正常生理状态造成明显影响。

汇报人:张子妍
审核:代一冯、任建君
