



精读分享│【Science translational medicine】:TP53/CDKN2A双基因敲除胃食管交界处类器官模型的构建及多组学分析
英文题目:Generation and multiomic profiling of a TP53/CDKN2A double-knockout gastroesophageal junction organoid modelTP53/CDKN2A
中文题目:TP53/CDKN2A双基因敲除胃食管交界处类器官模型的构建及多组学分析
期刊:Science translational medicine(IF: 15.8)
发表时间:2022年11月
PMID: 36449602
PMCID: PMC10026384
DOI: 10.1126/scitranslmed.abq6146

发表单位
美国南加州大学赫尔曼·奥斯特罗牙科学院颅面分子生物学中心兼诺里斯综合癌症中心,加利福尼亚州洛杉矶市,邮编90033。
美国约翰斯·霍普金斯大学医学院内科及肿瘤科胃肠病与肝病学系、西德尼·金梅尔综合癌症中心,马里兰州巴尔的摩市,邮编21287。
中国陕西省西安市雁塔西路277号,西安交通大学第一附属医院检验科,邮编710061。
美国宾夕法尼亚州费城市,爱因斯坦医疗网络,邮编19136。
美国加利福尼亚州洛杉矶市,西达-赛奈医学中心内科,邮编90048。
美国约翰斯·霍普金斯大学医学院,拉塞尔·H·摩根放射学与影像科学系癌症影像研究部,马里兰州巴尔的摩市,邮编21287。
美国约翰斯·霍普金斯大学医学院,西德尼·金梅尔综合癌症中心,马里兰州巴尔的摩市,邮编21287。
美国约翰斯·霍普金斯大学医学院,生物化学系,马里兰州巴尔的摩市,邮编21287。
美国约翰斯·霍普金斯大学医学院,病理学系,马里兰州巴尔的摩市,邮编21287。
中国陕西省西安市,西安交通大学第一附属医院转化医学中心,邮编710061。
研究流程图

摘要
在胃食管交界处(GEJ)肿瘤发生早期,肿瘤抑制基因肿瘤蛋白 p53(TP53)和细胞周期蛋白依赖性激酶抑制剂2A(CDKN2A)就会失活。然而,由于缺乏 GEJ 特异性疾病模型,TP53和CDKN2A 在 GEJ 失活所导致的促癌后果尚未得到明确。在此,研究者开发了野生型原代人 GEJ 类器官模型和 CRISPR 编辑的癌变转化 GEJ 类器官模型。在 GEJ 类器官中通过 CRISPR-Cas9 介导的 TP53 和 CDKN2A 敲除(TP53ko/CDKN2Ako)在体外诱导了形态学上的异型增生和癌前特征,并在体内形成了肿瘤。脂质组学分析发现,几种血小板活化因子(PTAFs)在 CRISPR 编辑的类器官中是上调最显著的脂质。通过 siRNA 敲低或药物抑制剂(WEB2086)消除 PTAF/PTAF 受体(PTAFR)可减少 TP53/CDKN2Ako GEJ 类器官在体外的增殖和其他癌前特征,并抑制体内肿瘤形成。此外,WEB2086 还抑制了 Eso26(一种已建立的人食管腺癌细胞系)的小鼠异种移植瘤。从机制上讲,TP53/CDKN2A 双重失活破坏了转录组和 DNA 甲基化组,这可能是由关键转录因子介导的,特别是叉头盒蛋白 M1(Forkhead box M1,FOXM1)。FOXM1 通过与 PTAFR 启动子结合激活 PTAFR 转录,进一步放大了 PTAF-PTAFR 通路。总之,这些研究建立了一个强大的模型系统,用于研究早期GEJ癌变性事件,确定了 GEJ 模型肿瘤发生过程中发生的代谢和表观基因组的关键变化,并揭示了一种潜在的癌症治疗策略。这项工作为早期 GEJ 肿瘤中与 TP53/CDKN2A 失活相关的癌变机制提供了见解,这可能有助于早期诊断和预防 GEJ 肿瘤。
背景
胃食管癌每年导致超过 100 万人死亡,是全球癌症死亡的第二大原因。其中一类肿瘤,即胃食管交界处(GEJ)腺癌,在美国和其他西方国家近几十年来的发病率增加了 2.5 倍以上(1, 2)。与胃食管区域的其他癌症相比,GEJ 肿瘤尤其具有侵袭性,预后极差;因此,迫切需要对其分子基础进行深入研究。然而,由于缺乏具有生物学相关性的 GEJ 特异性早期疾病模型,这一目标的实现一直困难重重。人类类器官是强大的模型,能够重现并维持其来源组织的基本遗传、功能和表型特征(3)。类器官填补了转基因动物模型和细胞系之间的知识空白,为理解基本的致癌机制和识别潜在的癌症治疗靶点提供了宝贵的平台(4)。
脂质代谢重编程是癌症的一个已知特征(5, 6),某些脂质谱已被推荐作为早期癌症检测的生物标志物或癌症治疗的靶点(7-9)。脂质组的命运和组成异常通过激活致癌途径和过程促进肿瘤发生,增强癌细胞生长,并支持癌细胞在具有挑战性的微环境中存活(9)。通过在胃食管癌中进行脂质组学分析,研究者们最近证明了特定脂质种类的合成增强对于癌细胞的存活和增殖是必需的(10, 11)。然而,胃食管交界处癌症脂质组的代谢变化在很大程度上仍不清楚。
在本研究中,研究者通过建立首个源自正常人GEJ的类器官,并利用 CRISPR-Cas9 基因编辑技术对其进行肿瘤蛋白 p53(TP53)和细胞周期蛋白依赖性激酶抑制剂 2A(CDKN2A)双基因敲除(TP53KO/CDKN2AKO )改造,来探究 GEJ 癌变的发生机制。借助这一 GEJ 特异性疾病模型,研究者探讨了 GEJ 细胞早期肿瘤性转化过程中的表型、代谢和表观基因组变化,旨在为这种侵袭性强且知之甚少的恶性肿瘤的发病机制提供见解。
结果
1、人正常胃食管连接部类器官的建立与表征
为解决缺乏具有生物学相关性的胃食管连接部(GEJ)特异性疾病模型的问题,研究者从人类内镜下获取的原代GEJ活检样本中生成了三维(3D)类器官,病理学检查确认这些样本中不含异型增生细胞和肿瘤细胞(表S1)。简而言之,新鲜分离的GEJ淋巴滤泡被嵌入基质胶中,并与含有关键干细胞生长因子的条件培养基一起培养(图1A和表S2)。在这些培养条件下,研究者成功地从六个正常GEJ活检样本中建立了3D类器官,成功率达到了100%。
研究者通过相差显微镜成像、苏木精-伊红(H&E)染色和细胞活力[水溶性四唑盐(WST-1)]检测对这些类器官进行了表征(图1B至D)。在初始接种后的第4天,形成了3D球状结构,直径达到25微米。这些结构持续生长,最终在第24至29天达到106微米的大小平台期(图1B和C)。这些GEJ类器官在第24天时由120至250个细胞组成,表明培养中的细胞倍增时间为77.85±5.54小时。
组织学分析显示,正常的胃食管连接处类器官形成了由单层上皮细胞构成的三维结构。类器官的细胞活力在第14天达到峰值,并在第24天显著(P<0.05)下降(图1D)。研究者持续监测了这六个独立的正常胃食管连接处活检样本的类器官;它们在体外连续增殖了4至6个月。
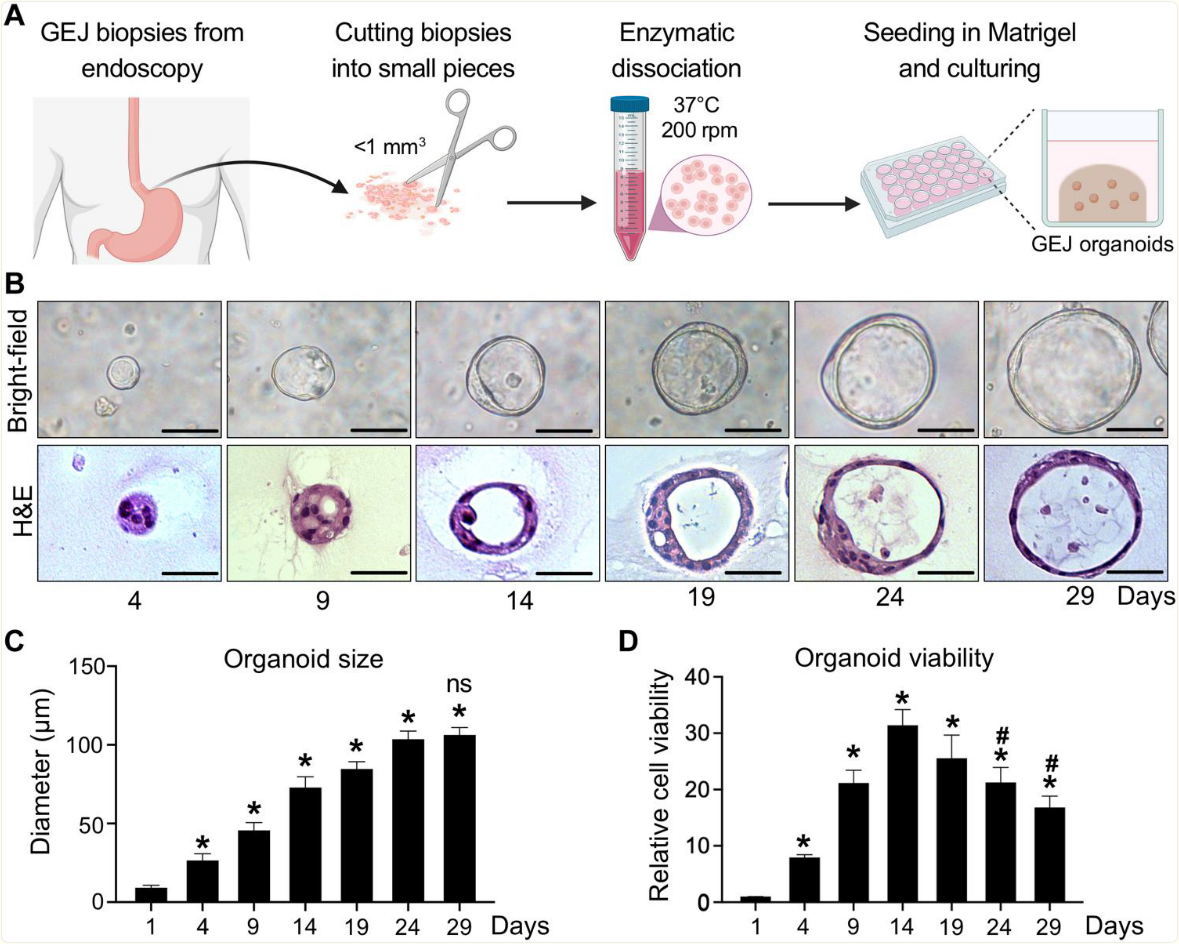
图 1. 人正常胃食管连接部类器官的建立与表征。
(A)从人类GEJ内镜活检标本生成类器官的工作流程。通过上消化道内镜获取正常 GEJ 黏膜活检标本,然后将其切碎并用酶消化。将细胞悬液与基质胶混合,在条件培养基中启动三维类器官培养。(B 至 D)在指定时间点对 GEJ 类器官的结构和生长特性进行分析。在相差显微镜下对三维类器官进行显微摄影(B,上)并收集用于苏木精 - 伊红(H&E)染色(B,下)。比例尺,50 微米。在每个时间点测定平均类器官大小(C)和存活率(D)。数据以平均值 ± 标准差表示;n = 6 个生物学重复样本。*P < 0.05 与第 1 天相比;#P < 0.05 与第 14 天相比;与第 24 天相比无显著差异(ns),通过方差分析。
2、TP53/CDKN2A 缺失促进胃食管结合部类器官的增殖、异型增生和肿瘤性转化
接下来,研究者试图构建一种GEJ癌变的疾病和病理模型,该模型能够更旺盛地生长,在培养中存活更久,并且更有利于对GEJ癌变转化的研究。由于TP53和CDKN2A是人类GEJ癌症中最常失活的两个肿瘤抑制基因(12–14),研究者选择使用CRISPR-Cas9基因组编辑系统在GEJ类器官模型中失活这两个基因。
研究者利用结合了CRISPR RNA(crRNA)和反式激活crRNA(tracrRNA)的gRNA复合物与Cas9核酸酶,制备了同时靶向TP53(外显子4)和CDKN2A(外显子1α)的Cas9:向导RNA(gRNA)核糖核蛋白(RNP)复合物。然后,研究者将人类GEJ类器官解离成每个由5至15个细胞组成的小细胞簇,并使用优化的方案通过电穿孔法将RNP复合物导入(图S1A)。
对于对照类器官组,研究者电穿孔导入了一个阴性对照的非靶向RNP复合物。电穿孔效率达到了42%(图S1B)。随后,研究者利用Nutlin-3a选择具有突变型TP53的类器官,Nutlin-3a可抑制小鼠双微体2(MDM2原癌基因),从而导致TP53野生型细胞生长停滞(图S1C)(15)。
选择完成后,研究者进行了Sanger测序以验证靶向TP53和CDKN2A外显子的特异性编辑。在TP53和CDKN2A靶位点观察到移码突变,包括1个碱基对(bp)的插入或缺失(图2A和B),这证实了GEJ类器官的基因组编辑成功。
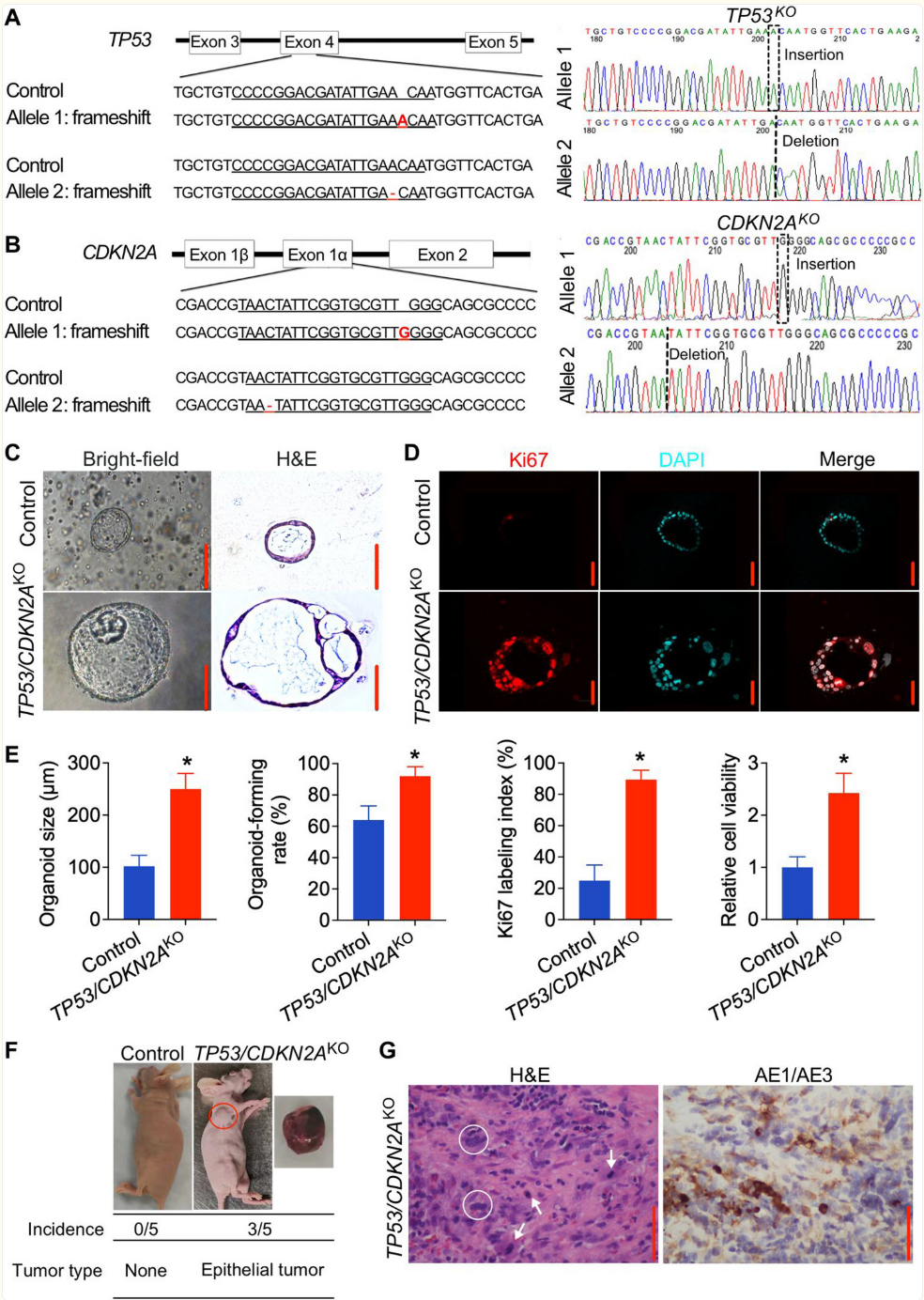
图 2. TP53/CDKN2A 基因敲除促进人类正常胃食管连接部类器官的肿瘤性转化。
(A 和 B)对 TP53/CDKN2A GEJ 类器官进行Sanger测序,显示 TP53(A)或 CDKN2A(B)中存在 1 个碱基的插入或缺失。红色字体表示基因组 DNA 中相应的移码插入缺失。(C 和 D)在接种 1×105悬浮的类器官细胞 10 天后,使用相差显微镜对类器官培养物进行显微摄影,并收集用于(C)明场、H&E 染色和(D)Ki67(红色)免疫荧光染色。DAPI,4',6-二脒基-2-苯基吲哚。(E)通过测量超过 50 个类器官来确定平均类器官大小、类器官形成效率和 Ki67 指数。数据以平均值±标准差表示;n = 4 个生物学重复。*P < 0.05,通过学生 t 检验。(F)用对照或 TP53/CDKN2Ako GEJ类器官注射小鼠后形成的异种移植物的代表性图像;下方表格显示了发病率和肿瘤特征。该实验重复了一次,结果相似。(G)由 TP53/CDKN2Ako 类器官产生的异种移植物的代表性 H&E 和 AE1/AE3 全角蛋白免疫组化染色(棕色)。白色箭头,有丝分裂;白色圆圈,异常大、多形性细胞,核膜不规则;比例尺,100 微米。
接下来,研究者对GEJ类器官在TP53和CDKN2A缺失后的表型变化进行了表征。在接种后第10天,对照类器官形成了单层上皮细胞,细胞核正常。相比之下,TP53/CDKN2A缺失的类器官直径更大,结构更复杂,具有增强的有丝分裂活性,细胞核更大且出现不典型形态。这些改变与不典型增生的组织学特征一致(图2C和D)(16)。
此外,研究者对三叶因子3(TFF3)进行了免疫荧光染色。TFF3是肠上皮化生的候选诊断标志物,结果显示在TP53/CDKN2A缺失的GEJ类器官中TFF3表达水平升高(图S2)。这一发现表明TP53/CDKN2A的失活可能促进GEJ柱状上皮细胞向肠上皮化生方向转化。
TP53/CDKN2A缺失的类器官其形成效率也明显高于对照组(92%对64%,P<0.05;图2E)。免疫荧光染色进一步显示,TP53/CDKN2A缺失类器官的Ki67标记指数显著上升(89.4%对24.9%;图2D和2E)。WST-1检测结果显示,TP53/CDKN2A缺失类器官的增殖能力为对照类器官的2.4倍(图2E)。
此外,在相同培养条件下,对照类器官的最长维持时间为6个月,而TP53/CDKN2A缺失类器官可连续培养超过19个月,其估算群体倍增时间为37.86±1.90小时(图S3)。这些数据表明,TP53/CDKN2A缺失能够增强GEJ类器官在体外的持续增殖能力和异型增生特征。
为了评估TP53/CDKN2A失活在胃食管结合部类器官中的体内效应,研究者进行了异种移植实验。将对照组和TP53/CDKN2A失活的类器官细胞(每次注射2×10^5个细胞)分别皮下注射到五只裸鼠的左、右腋窝。在注射后的5个月观察期内,注射对照组胃食管结合部类器官的五只裸鼠均未形成肿瘤。相反,注射TP53/CDKN2A失活类器官的五只裸鼠中有三只在8周内形成了肿瘤(图2F)。这些肿瘤在形态上类似于高度分化的胃食管腺癌。对源自TP53/CDKN2A失活类器官的异种移植物进行H&E和免疫组化(IHC)分析,发现有增生现象,细胞异常增大、多形性,核膜不规则,并且抗细胞角蛋白1抗体(AE1)/AE3蛋白呈阳性表达(图2G)。因此,这些体内数据表明TP53/CDKN2A失活直接促进了胃食管结合部类器官的肿瘤转化。
3、脂质组学基质辅助激光解吸电离成像质谱法(MALDI-IMS)发现磷脂酰甘油酰胺(PTAFs)是 TP53/CDKN2A 缺失类器官中上调最显著的脂质
脂质代谢的重新编程和失调是癌症的一个显著特征(17)。然而,尚不清楚在早期胃食管交界处癌变过程中脂质代谢过程是否以及如何发生改变。为了解决这一知识空白,研究者应用脂质组学基质辅助激光解吸电离成像质谱(MALDI-IMS)技术,以2,5-二氢苯甲酸(DHB)作为基质,在正离子模式下对TP53/CDKN2A KO与对照类器官进行分析,以发现磷脂等脂质种类的变化(18)。
在质量荷比(m/z)40至2000的质量范围内,研究者通过直接分析类器官切片获得了脂质种类的质量谱(图S4A),而每个峰的离子图像则显示了在类器官切片中的位置和相对强度(图S4B)。在成像实验中,研究者设定m/z > 450的阈值,以减少背景基质信号,并专注于通常大于m/z 400的磷脂。
研究者首先基于单个峰的平均强度数据的倍数变化进行分析,在TP53/CDKN2A KO对比对照类器官的平均倍数变化大于1.5的情况下,鉴定出50个上调峰和132个下调峰(图3A和表S3)。同时,研究者进行了受试者工作特征(ROC)分析,采用曲线下面积(AUC)阈值大于0.75。以此标准,TP53/CDKN2A KO对比对照类器官中分别有16个和50个峰上调和下调(图3B和表S4)。一致地,通过ROC方法鉴定出的所有这些改变的峰,其倍数变化方向相同(图3B)。
接下来,研究者在类器官上进行了串联质谱(MS/MS)(图S5),并鉴定出在16个共享上调峰中,AUC最高的峰(m/z = 467.20)是一种特定的血小板活化因子(PTAF)脂质PC-O-14:0(LMGP01020009)。此外,还鉴定出另外四种脂质为PTAF脂质[m/z 451.16,PC-O-13:1(LMGP01020146);m/z 550.03,PC-O-20:0(LMGP01020094);m/z 549.06,PC-O-20:1(LMGP01020146)]或者是一种PTAF脂质的前体[m/z 482.19,LPC-O-16:0(LMGP01060010)]。这些脂质的MALDI成像数据和化学结构显示在图3(C和D)中。PTAF脂质是一类甘油磷胆碱,被认为在多种病理过程中作为生物活性介质发挥作用,包括肿瘤血管生成和转移(19,20)。
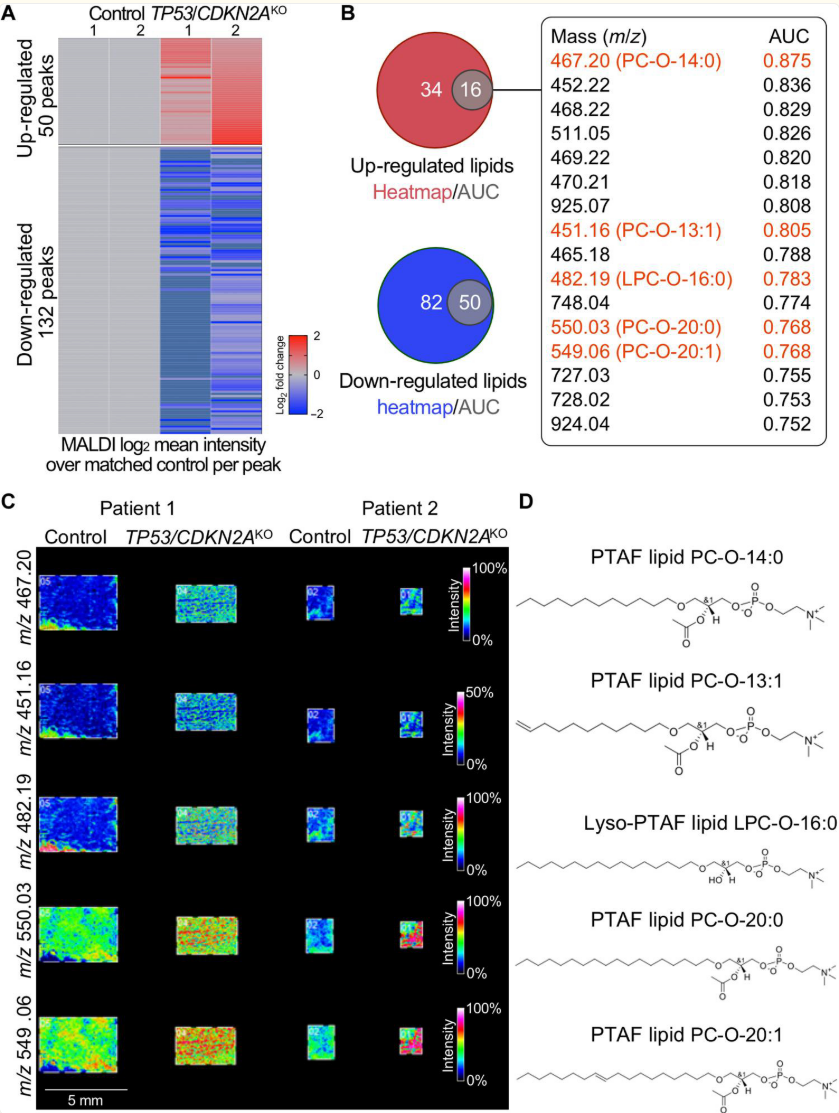
图 3. 与对照组胃食管连接部类器官相比,TP53/CDKN2A KO 中的 PTAF 脂质增加。
基于基质辅助激光解吸电离成像的脂质组学分析在来自两名不同患者的独立配对的 TP53/CDKN2A 缺失型与对照型胃食管结合部类器官中进行。图(A)为具有 m/z 值大于 450、平均绝对倍数变化值大于1.5且每个 TP53/CDKN2A 缺失型类器官中个体绝对倍数变化大于1.2的鉴别脂质峰(m/z)的热图,与匹配的对照组相比。图(B)为基于 AUC 和热图结果的上调和下调脂质重叠的维恩图。列出了16种重叠上调脂质及其AUC值。图(C)为TP53/CDKN2A缺失型类器官中代表性PTAF脂质的MALDI成像,图(D)为其相应的化学结构。
4、阻断 PTAF/PTAFR 可抑制 TP53/CDKN2A 缺失型胃食管结合部类器官的肿瘤特性
在鉴定出多个PTAF脂质为TP53/CDKN2AKO GEJ类器官中增加的磷脂后,研究者探讨了它们在GEJ癌变发生中的潜在作用。作为甘油磷胆碱,PTAF通过与其同源受体PTAFR结合发挥生物学效应(21)。因此,研究者首先评估了GEJ癌变中PTAFR的表达,包括研究者的类器官模型和癌症基因组图谱(TCGA)数据集。与其同源脂质配体相似,TP53/CDKN2A KO组织相对于对照组织中PTAFR的表达显著上调(P<0.05;图4A)。与这一发现一致的是,TCGA食管腺癌(EAC)样本的PTAFR mRNA表达高于非恶性TCGA GEJ样本(图S6)。
受这些观察结果的鼓舞,研究者接下来在早期GEJ模型系统中直接通过小干扰RNA(siRNA)敲低或药物抑制来消除PTAF/PTAFR功能。通过siRNA沉默PTAFR表达显著(P<0.05)降低了TP53/CDKN2AKO组织的平均大小、细胞活力和Ki67指数(图4,B至E)。同时,研究者用载体对照(0.1%二甲基亚砜(DMSO))或特定的PTAFR药理拮抗剂WEB2086(不同浓度)处理TP53/CDKN2AKO类器官。与siRNA结果一致,WEB2086处理后的TP53/CDKN2A KO类器官大小显著减小(P<0.05),Ki67指数也显著降低(图4F至H)。WST-1检测显示,从第4天开始,代谢活跃细胞开始减少,并且在第7天和第10天确认了时间和剂量依赖性的抑制作用(图4H)。
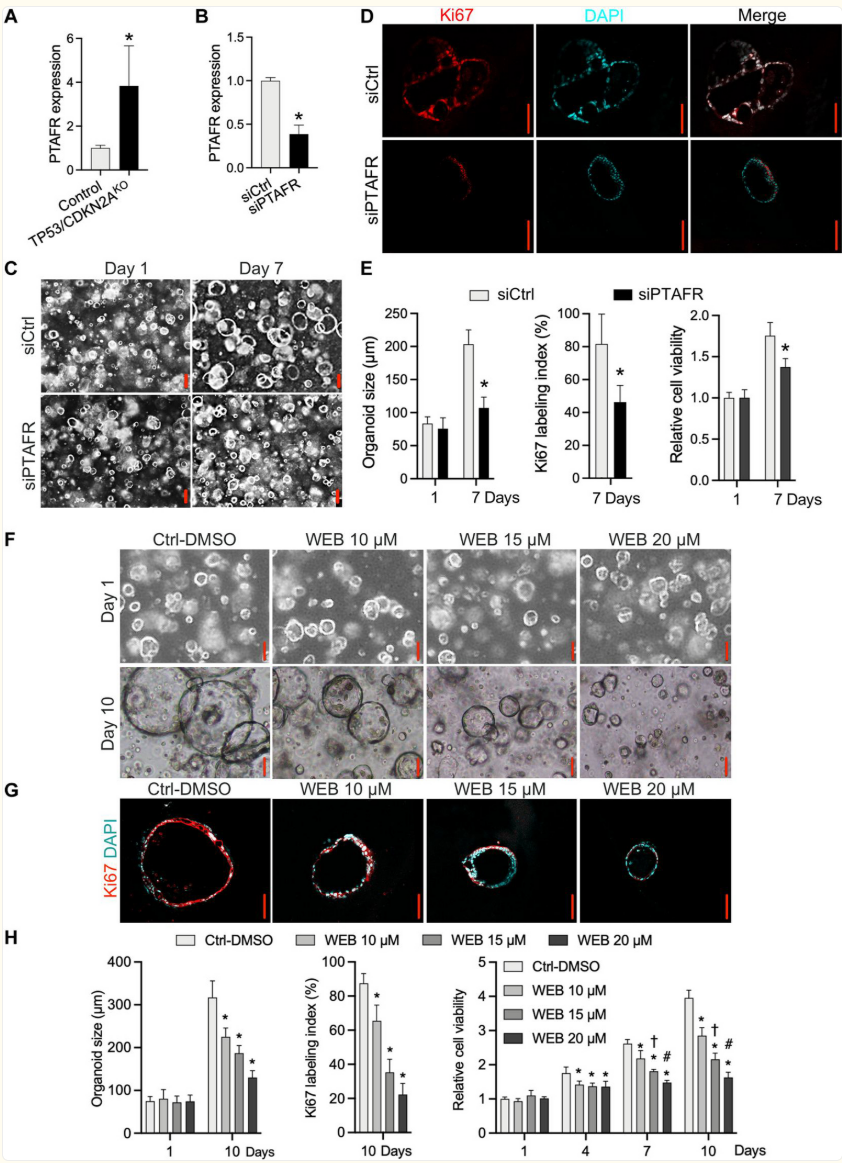
图 4. 阻断 PTAF/PTAFR 可抑制 TP53/CDKN2A 缺失型胃食管结合部类器官的生长和增殖。
(A)TP53/CDKN2AKO与对照组GEJ类器官中PTAFR的mRNA表达情况。(B)通过siRNA敲低TP53/CDKN2AKO GEJ类器官中PTAFR的mRNA。(C)将用对照沉默RNA(siCtrl)和PTAFR沉默RNA(siPTAFR)处理的类器官在相差显微镜下拍照,并收集用于(D)Ki67免疫荧光染色。(E)在指定时间点定量平均类器官大小和Ki67标记指数,并通过WST-1测定法测定细胞活力。*P < 0.05 与同一天的siCtrl相比。比例尺,100 μm。(F 至 H)TP53/CDKN2A KO GEJ类器官用溶剂对照(0.1% DMSO)或特定PTAFR药理拮抗剂WEB2086在不同浓度下处理。通过相差成像和WST-1测定法分别测定平均类器官大小(F)和Ki67免疫荧光图像(G)以及细胞活力(H)。(G)在第10天获得Ki67标记图像和定量(H)。比例尺,100 μm。数据以平均值±标准差表示;n = 4个生物学重复。*P < 0.05 与同一天的Ctrl-DMSO相比;†P < 0.05 与同一天的WEB 10 μM相比;#P < 0.05 与同一天的WEB 15 μM相比,通过方差分析。
接下来,研究者在体内测试了对血小板活化因子受体(PTAFR)的抑制作用。将TP53/CDKN2A缺失的类器官细胞(每注射2×10⁵个细胞)皮下注射到裸鼠腋窝。接种3天后,每隔2天通过腹腔注射给予对照剂(1.25%DMSO)或WEB2086(5毫克/千克/天),持续3周。
在3个月的注射后观察期内,对照组(DMSO)的5只注射小鼠中有3只在7周内形成了肿瘤(图5A),这与研究者上面观察到的情况相似(图2F)。WEB2086治疗则完全阻止了TP53/CDKN2A缺失类器官的肿瘤形成(图5A)。
接下来,研究者在源自已建立的食管腺癌(EAC)细胞系(Eso26)的异种移植模型中评估了WEB2086的效果。同样,PTAFR抑制显著抑制了EAC异种移植瘤的生长和增殖(P<0.05;图5B和C)。免疫组化分析显示,WEB2086处理的Eso26肿瘤异种移植瘤中Ki67的表达下调(图5D)。
这些结果表明,阻断PTAF/PTAFR脂质级联反应能显著抑制TP53/CDKN2A KO GEJ类器官在体外和体内的生长、增殖和肿瘤发生,提示PTAF/PTAFR在介导胃食管连接部早期肿瘤进展中具有重要作用。
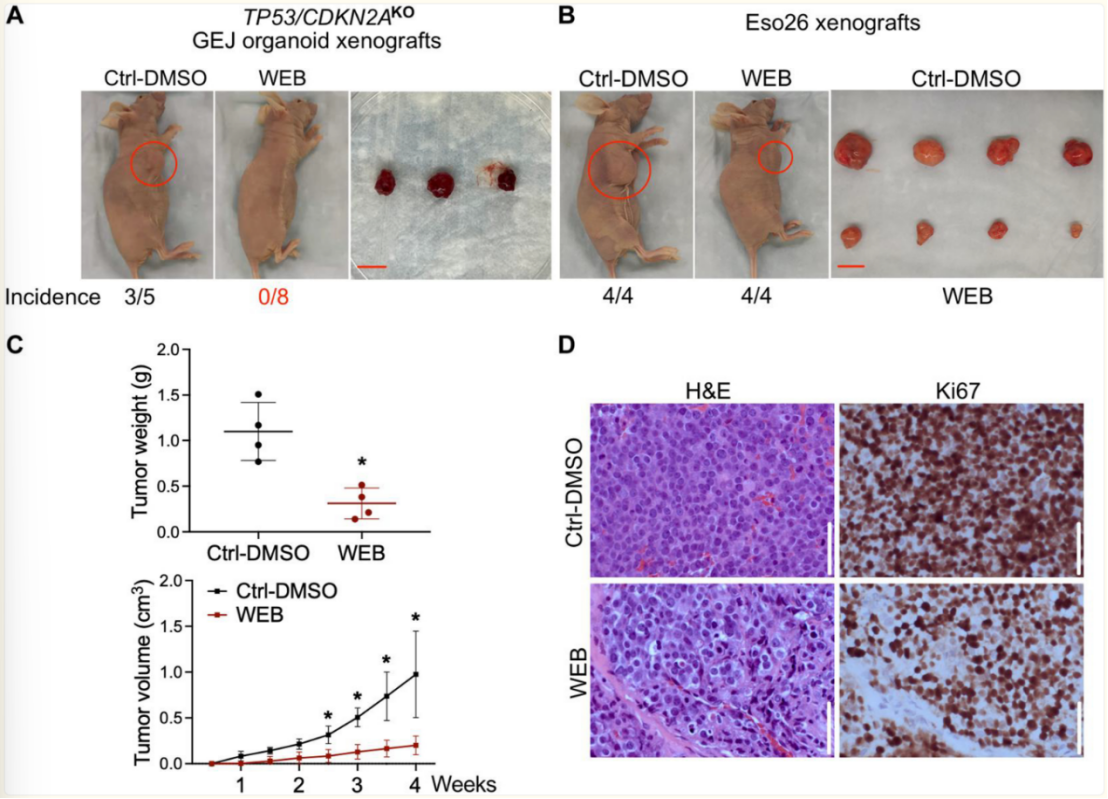
图 5. 阻断PTAF/PTAFR通路可抑制TP53/CDKN2AKO GEJ 类器官和Eso26细胞在体内的肿瘤发生。
将TP53/CDKN2AKO组织细胞或Eso26细胞(每次注射2×10⁶个细胞)皮下注射至裸鼠腋窝。接种3天后,每2天通过腹腔注射给予WEB2086(5毫克/千克/天)或溶媒对照(1.25%DMSO溶于PBS中),持续3周。(A)WEB2086治疗后TP53/CDKN2AKO GEJ组织细胞的异种移植图像及发病率。比例尺,1厘米。该实验重复一次,结果一致。(B)WEB2086治疗后Eso26异种移植瘤的外观图像及发病率。比例尺,1厘米。(C)第4周肿瘤重量及WEB2086处理与否对Eso26异种移植瘤生长的影响。数据以平均值±标准差表示;n = 5。*P < 0.05,与同一时间点的Ctrl-DMSO组比较,采用Student t检验。(D)Eso26异种移植瘤的H&E染色与Ki67免疫组化图像。比例尺,100微米。
5、TP53/CDKN2A失活会改变胃食管结合部类器官的 DNA 甲基化组和转录组
为了探究对照组与TP53/CDKN2A缺失类器官在转录组和表观基因组层面的差异,研究者分别进行了转录组测序(RNA测序,RNA-seq)和Illumina MethylationEPIC芯片分析。具体而言,对来自四名患者的配对野生型和TP53/CDKN2A缺失类器官进行了RNA-seq。
与对照组相比,TP53/CDKN2A缺失类器官中存在556个差异表达基因(DEGs)显著不同(校正P值<0.05且绝对倍数变化>2;图6A和表S5),其中312个上调,244个下调。对这些基因进行基因本体论(GO)分析,发现与有丝分裂进入和细胞周期进程相关的生物学过程和通路显著富集(图6B和表S6)。这一发现与上述TP53/CDKN2A缺失类器官显著加速增殖和生长的观察结果一致。

图 6. TP53/CDKN2AKO GEJ类器官的DNA甲基化组和转录组分析。
(A)火山图展示了 TP53/CDKN2AKO类器官与对照类器官中差异表达基因(DEGs)的情况。数据代表了四个生物学重复样本。(B)基因本体论(GO)分析确定了TP53/CDKN2AKO类器官与对照类器官中差异表达基因(DEGs)在生物过程和细胞成分方面排名靠前的关键术语。(C)火山图展示了 TP53/CDKN2AKO 类器官与对照类器官中差异甲基化的 CpG(红色表示对照组中高甲基化,蓝色表示TP53/CDKN2AKO组中高甲基化)。(D)柱状图展示了 TP53/CDKN2AKO类器官与对照类器官中差异甲基化区域(DMRs)的情况。(E)在TP53/CDKN2AKO类器官与对照类器官中低甲基化的差异甲基化区域(DMRs)中富集的来自叉头框(FOX)家族的排名靠前的转录因子(TF)结合基序。P 值使用 HOMER 软件包计算得出。P1,患者 1;P2,患者 2;P3,患者 3。(F)点图展示了TP53/CDKN2AKO类器官与对照类器官(底部点,n = 4 个生物学重复样本)以及来自 TCGA 的食管腺癌(EAC)肿瘤(n = 88)与正常GEJ样本(n = 9)中 FOX 家族转录因子(TFs)的表达情况(顶部点)。基因是根据比较食管胃结合部腺癌(EAC)肿瘤与正常食管胃结合部(GEJ)样本的 P 值的-log10值进行排序的。点的大小表示-log10(P 值);颜色表示log6 倍变化;未显示的点对应于未检测到的 mRNA 表达。
在表观基因组水平上,研究者确定了来自每位患者的对照组和TP53/CDKN2A KO组织类器官之间的差异甲基化CpG位点和差异甲基化区域(DMR)。例如,在来自患者1的组织类器官中,突变型和对照组组织类器官中分别有1732个和1391个CpG二核苷酸显著低甲基化(校正P<0.05且绝对甲基化变化量>0.2)(图6C)。这些CpG位点分别对应于TP53/CDKN2AKO和对照组组织类器官中的129个和83个低甲基化DMR(图6D和表S7)。来自其他三位患者的组织类器官的结果见图S7和表S7。
已知DNA低甲基化区域包含与转录因子(TFs)结合相关的调控元件(22,23)。为了确定在研究者的GEJ癌变类器官模型中涉及的候选转录因子,研究者使用超几何优化基序富集(HOMER)软件包研究了低甲基化差异甲基化区域(DMRs)中富集的转录因子识别基序序列(24)。在TP53/CDKN2AKO类器官的低甲基化DMR中,叉头框(FOX)转录因子家族的基序是富集程度最高的序列之一(图6E)。
由于不同FOX转录因子家族成员识别相似的基序序列(25),研究者接下来试图确定哪些因子参与其中。先前的研究表明,特定转录因子的表达与其占据区域的去甲基化程度相关(26,27)。因此,研究者基于研究者类器官的RNA-seq数据以及来自TCGA的正常GEJ和食管腺癌(EAC)数据,分析了26种FOX转录因子的mRNA表达。FOXM1和FOXC2在TP53/CDKN2AKO类器官中均有所增加。FOXM1在EAC组织与正常GEJ组织相比也呈上调状态(图6F),这表明其可能是低甲基化DMRs中富集的候选转录因子,在TP53/CDKN2AKO类器官中可能具有增强的活性。FOXM1是已知的癌症细胞增殖和细胞周期进程的调节因子(28,29),这与研究者在TP53/CDKN2AKO类器官中观察到的癌前表型相符。
6、PTAFR是FOXM1的直接转录靶点
研究者上述的数据不仅将磷脂酰血小板活化因子(PTAF)磷脂类分子确定为在脂质组学分析中诱导程度最高的脂质分子类别之一,还揭示了其同源受体 PTAFR在TP53/CDKN2AKO的GEJ类器官中的表达上调(图 4)。为了进一步阐明TP53/CDKN2A缺失类器官中PTAFR表达上调的机制,研究者对PTAFR启动子进行了基序富集分析,结果显示FOX基序序列再次高度富集(第4项)(图7A)。鉴于研究者上述数据已将FOXM1确定为可能参与TP53/CDKN2A缺失类器官表观遗传改变的调节因子,该分析具有相关性。
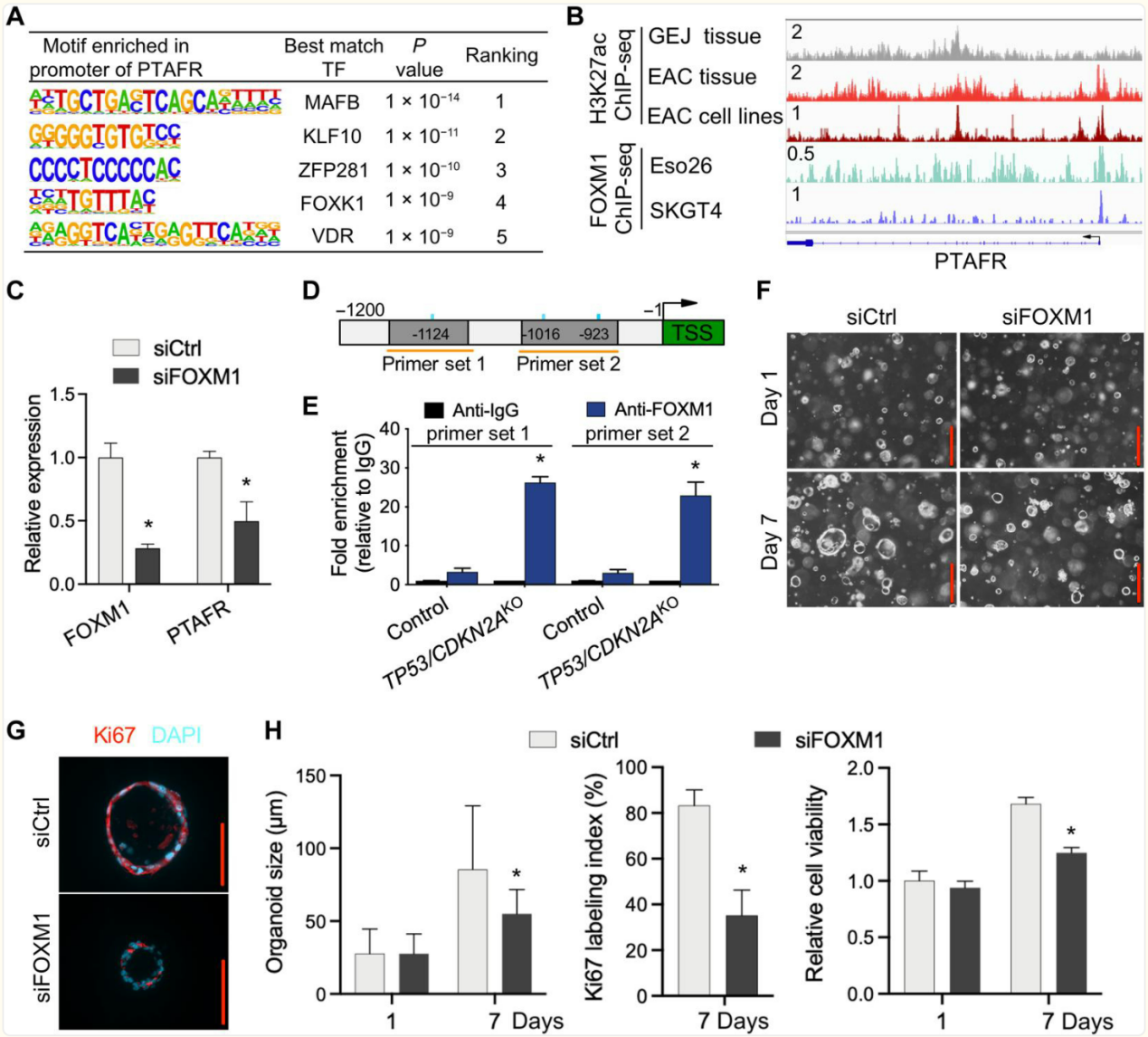
图 7. PTAFR 是 FOXM1 的直接下游靶点。
(A)类器官中PTAFR启动子区域的基序富集分析。(B)在指定组织和细胞系中PTAFR位点的H3K27ac和FOXM1的ChIP-seq图谱。所有H3K27ac信号均以相同比例显示。(C)在TP53/CDKN2AKO GEJ类器官中,用siRNA敲低FOXM1后PTAFR mRNA的相对表达量。(D)PTAFR转录起始位点(TSS,绿色)及其上游序列示意图。用于ChIP实验的PCR引物(橙色水平线)和FOXM1结合基序(蓝色垂直线)已标明。(E)使用(D)中所示引物对,在类器官中进行PTAFR启动子区域的FOXM1占据情况的ChIP-qPCR检测。使用抗FOXM1或抗IgG抗体进行ChIP,以抗IgG为参照计算相对富集倍数。数据以平均值±标准差表示;n = 4个生物学重复。*P < 0.05,与对照组比较,通过方差分析。(F)用siCtrl和siFOXM1处理的类器官在相差显微镜下拍照,并收集用于(G)Ki67免疫荧光染色。(H)量化平均类器官大小和Ki67标记指数,并通过WST-1检测细胞活力。比例尺,100微米。数据以平均值±标准差表示;n = 4个生物重复样本。* 与同一天的siCtrl组相比,P < 0.05(通过方差分析得出)。
为了验证FOXM1是否调控PTAFR的转录,研究者首先使用抗FOXM1抗体进行了染色质免疫沉淀测序(ChIP-seq)。在两种不同的食管腺癌(EAC)细胞系中,FOXM1占据了PTAFR的启动子区域(图7B)。此外,研究者最近的H3K27ac ChIP-seq数据(30)显示,在EAC原发肿瘤和细胞系中,PTAFR的启动子和候选增强子区域均具有强烈的H3K27ac信号(图7B),表明其转录活性很强。相反,在正常GEJ样本中,H3K27ac在PTAFR位点几乎未沉积,表明转录活性较弱或处于抑制状态(图7B)。这种组蛋白修饰模式与PTAFR的mRNA表达一致,其在EAC样本中相对于正常GEJ样本上调(图S6)。
由于上述ChIP-seq数据是在食管胃结合部腺癌(EAC)细胞系和原发肿瘤中生成的,因此研究者接下来在GEJ类器官中进行了FOXM1的染色质免疫沉淀定量聚合酶链反应(ChIP-qPCR)。在TP53/CDKN2A缺失的类器官中,内源性FOXM1在PTAFR启动子处显示出显著的占据,其富集程度比野生型GEJ类器官高出7.9倍以上(图7D和E)。为了验证FOXM1对PTAFR转录的调控作用,研究者在TP53/CDKN2AKO的类器官中使用siRNA沉默FOXM1,结果显示FOXM1敲低显著(P<0.05)降低了PTAFR的表达(图7C)。
在TP53/CDKN2A缺失的类器官中,FOXM1沉默降低了平均类器官大小、细胞活力和Ki67指数(图7F至H),这与研究者通过PTAF/PTAFR阻断所获得的效果一致。此外,研究者还测量了正常对照、TP53缺失和TP53/CDKN2A缺失类器官中PTAFR和FOXM1的mRNA表达(图S8)。这些实验表明,TP53单基因敲除增加了GEJ类器官中PTAFR和FOXM1 mRNA的表达,而TP53/CDKN2A双基因敲除则进一步放大了这种效应。因此,这些数据表明,TP53/CDKN2A失活增强了FOXM1与PTAFR启动子的直接结合,从而增加了GEJ类器官中PTAFR的表达和增殖。
讨论
理解GEJ癌变的分子起源和生物学特性的一个主要障碍在于缺乏合适的生物学相关模型,尤其是早肿瘤事件的相关模型。由正常上皮细胞衍生的原代三维类器官培养物是研究原始天然组织体外关键特性(包括形态学、组织学和分子特征)的理想系统(3)。据研究者所知,研究者在此首次描述了源自正常胃食管结合部的人类类器官培养模型,并提供了一种高度可靠的方案,确保从内镜活检中成功培养类器官。研究者证明,野生型胃食管结合部类器官在体外可培养至少4个月,而TP53/CDKN2A缺失型类器官甚至可以培养更长时间(至少19个月)。该平台为模拟胃食管结合部相关疾病、表征健康和患病胃食管结合部状况以及寻找从正常到病理胃食管结合部转变的分子机制提供了巨大希望。
此外,研究者在此还证明,经CRISPR工程改造的人类TP53/CDKN2A缺失类器官为模拟GEJ早期肿瘤事件提供了一种有效的工具。在后来发展为高级别上皮内瘤变或EAC的良性巴雷特食管(BE)患者中检测到了TP53突变(31–33),这表明其参与了GEJ解剖区域的早期肿瘤形成。同样,超过80%的BE和EAC组织中CDKN2A失活(34–36)。类似的分子异常也出现在胃贲门肿瘤中(37–42),这支持了TP53和CDKN2A对涉及GEJ区域的多种腺癌发生有贡献。与这些静态观察结果一致,研究者的动态模型表明,TP53/CDKN2A失活直接导致了与GEJ癌变进展一致的生物学和分子特征。TP53和CDKN2A已被证实参与了多种脂质代谢过程(43-46)。然而,据研究者所知,目前尚无直接证据表明TP53和CDKN2A与GEJ癌变过程中出现的脂质代谢异常有关。在此,研究者证明了TP53/CDKN2A失活会直接改变GEJ类器官的脂质组学特征。研究者发现,在GEJ类器官中,由TP53/CDKN2A失活而上调的脂质主要包括几种PTAFs,这是一类具有已知信号传导作用的磷脂介质,在包括癌症发生在内的许多重要生物学过程中发挥着作用(21,47–50)。在猫食管炎模型中,PTAF磷脂水平升高(51);在胃癌患者中,PTAF磷脂水平升高的频率也高于健康对照组(52)。PTAFs主要通过与其特异性受体PTAFR结合发挥作用(53–55)。PTAFR在人类胃腺癌组织中大量表达;此外,PTAFR过表达与不良的临床预后参数密切相关(56–58)。与这些已知的临床和生物学数据一致,研究者在此确定PTAF/PTAFR是由TP53/CDKN2A KO在胃食管交界处诱导的肿瘤进展的病因性介质。
在临床前研究中,已有研究使用小分子抑制剂在多种体外培养的癌细胞中探讨了PTAF/PTAFR通路的靶向效果(59,60)。然而,由于类器官模型技术的限制,PTAF/PTAFR通路在早期肿瘤转化过程中的作用迄今仍未被系统研究。在本研究建立的GEJ类器官模型中,PTAFR拮抗剂WEB2086有效抑制了TP53/CDKN2AKO类器官中发生的早期肿瘤表型变化,并在Eso26食管腺癌细胞模型中也表现出类似的生长抑制效果。
研究者的数据进一步表明,TP53/CDKN2A失活可引发表观遗传与转录程序的重编程,推动正常GEJ组织向恶性转化。在本研究中,综合分析揭示FOXM1结合基序在低甲基化区域富集,且该转录因子在TP53/CDKN2A失活背景下显著上调。FOXM1已被广泛报道在多种实体瘤中过度表达(61–63),其下游信号通过与多个细胞通路的交叉调控促进癌症的发生与进展。本研究进一步证明,FOXM1可直接结合PTAFR启动子区,激活PTAFR的转录表达,形成功能性调控环路。与抑制PTAF/PTAFR通路所得结果一致,在TP53/CDKN2A缺失的GEJ类器官中敲低FOXM1同样可导致显著的细胞生长抑制。
研究者建立的人类原发性良性GEJ类器官模型,以及基于TP53/CDKN2A基因敲除诱导形成的癌前病变模型,为早期GEJ癌变的发生机制及其进展提供了一个可用于分子层面研究的有力平台。然而,本研究仍存在一定的局限性。体外类器官模型难以全面重现体内肿瘤发生过程中复杂的组织微环境和免疫系统参与;此外,研究者所使用的患者样本数量有限,可能影响结果的广泛适用性。未来可通过改进类器官培养系统(如引入器官芯片技术)以及扩大患者样本规模,进一步提升模型的生理相关性,并系统评估PTAF/PTAFR通路在GEJ癌变治疗中的潜在价值。
总之,研究者成功建立了一个强大且高度可控的类器官模型系统,可用于研究GEJ早期肿瘤发生的关键事件。通过脂质组学、表观遗传学和转录组学的综合分析,研究者揭示了PTAFs这一磷脂家族及其受体PTAFR在GEJ癌变转化过程中的促肿瘤作用。这一通路不仅为理解GEJ恶性转化机制提供了新视角,也代表了潜在的治疗干预靶点。研究者提出的将人类正常原发上皮类器官转化为具有肿瘤性特征的方法学框架,未来有望推广至其他癌症模型的构建,从而深化对肿瘤特异性基因调控网络的理解,推动新型靶向治疗策略的发展。
材料与方法
本研究旨在建立一个稳健的模型来探究早期GEJ的癌变转化过程,识别GEJ模型肿瘤发生过程中的关键事件,并发现潜在的癌症治疗策略。研究使用了患者来源的正常GEJ类器官,并利用CRISPR-Cas9基因编辑系统将其改造为TP53/CDKN2A缺失的类器官。在进行基因和化学扰动后,研究者对正常对照类器官与TP53/CDKN2A缺失类器官的表型、代谢、转录组和表观基因组变化以及肿瘤发生情况进行了研究。每个图例中均标明了重复次数。
患者样本
根据约翰斯·霍普金斯医院经批准的机构审查委员会方案,在患者书面知情同意的情况下,从约翰斯·霍普金斯医院接受诊断性食管胃十二指肠镜检查的患者胃食管连接部获取了主要的人类内镜活检样本。用于生成类器官的组织样本经病理学确认为正常的胃底腺-泌酸腺黏膜:未观察到杯状细胞或巴雷特黏膜特有的特殊上皮(表 S1)。
细胞系及其培养维护
L Wnt-3A细胞(CRL-2647)购自美国典型培养物保藏中心,用含10%胎牛血清(FBS)的杜尔贝科改良伊格尔培养基(DMEM)培养以制备Wnt-3A条件培养基。Cultrex HA-R-Spondin1-Fc 293T细胞(3710-001-01)购自Bio-Techne,用含10% FBS的DMEM培养以生成R-spondin-1条件培养基。Eso26细胞购自Jennio生物技术公司,用含10% FBS的RPMI 1640培养基培养。
胃食管交界处类器官培养
如图1A所示建立了GEJ类器官。简而言之,新鲜的内镜下GEJ活检样本保存在冰冷的条件培养基磷酸盐缓冲液(PBS)[含10μM Rho相关激酶(ROCK)抑制剂Y27632、2%(v/v)青霉素/链霉素和1×Primocin的PBS]中,直至24小时内进一步处理。用条件培养基PBS至少冲洗活检样本五次后,使用微型解剖剪刀将样本剪成小于1毫米的碎片。将组织碎片在含2.5%(v/v)胎牛血清、1%(v/v)青霉素/链霉素、胶原酶IX(1毫克/毫升)和II型分散酶(120微克/毫升)的DMEM中,在37°C下以200转/分钟的速度振荡消化40至90分钟。在4°C下以400g离心3分钟后,将沉淀的细胞团重悬于基质胶中。使用24孔板,每孔以50微升基质胶接种2000个细胞。在37°C下孵育10分钟使基质胶凝固后,向每孔加入500微升生长培养基。
胃食管结合部类器官的培养基为Advanced DMEM/F-12,添加了50%(体积比)自制的Wnt-3A条件培养基、20%(体积比)自制的R-spondin-1条件培养基、1%(体积比)青霉素/链霉素、10纳摩尔前列腺素E2、人成纤维细胞生长因子10(100纳克/毫升)、人表皮生长因子(50纳克/毫升)、Noggin(100纳克/毫升)、1毫摩尔N-乙酰半胱氨酸、10毫摩尔烟酰胺、10纳摩尔胃泌素I、500纳摩尔A-83-01、10微摩尔SB202190、10微摩尔Y27632、5微摩尔CHIR99021(仅用于最初的1至2次传代)、1×Primocin和1×B-27补充剂。培养基每3天更换一次,直至类器官准备好传代。
传代时,将类器官用PBS洗涤,然后在含有10微摩尔Y27632的TrypLE中消化5至7分钟,温度为37°C。孵育结束后,加入DMEM/F12终止消化。通过移液管机械分离类器官,然后在500g下离心3分钟。将沉淀重悬于基质胶中,每滴50至100微升的细胞基质胶悬液接种到新的培养板上。用于类器官培养的试剂列于表S2中。
类器官活力测定(WST-1 测定)
为了定量代谢活跃的活细胞,将类器官接种到 96 孔板中进行培养。在指定的时间点,向96孔板的每个孔中加入10微升细胞增殖试剂盒WST-1,并与类器官一起孵育90分钟。孵育结束后,仅将培养基转移到新的96孔板的孔中,用赛默飞世尔科技的酶标仪在450纳米波长下读取吸光度。所有实验均重复三次。
CRISPR-Cas9 基因组编辑技术在胃食管结合部类器官中的应用
类器官使用NEPA21(Nepa Gene)系统和Alt-R CRISPR-Cas9系统(Integrated DNA Technologies)进行电穿孔。Cas9:gRNA RNP复合物的制备方法如下:为制备100μM的gRNA复合物,将200μM的ATTO 550标记的tracrRNA和200μM的crRNA按等摩尔浓度混合,在95°C加热5分钟,然后缓慢冷却至室温;为每次电穿孔制备RNP复合物,将6μl的gRNA复合物(100μM)、8.5μg的Cas9核酸酶(10μg/μl)和10.5μl的双链缓冲液混合,在室温下孵育10分钟。RNP复合物在-80°C下保存以备后续使用。
电穿孔前两天,将类器官传代培养,并在不含抗生素的类器官培养基中培养,培养基中含5μM的CHIR99021。将类器官解离成10至15个细胞的团簇,用含4μM电穿孔增强剂的80μl电穿孔缓冲液重悬,然后与25μl靶向TP53的RNP复合物和25μl靶向CDKN2A的RNP复合物混合。混合物转移至预冷的2毫米电穿孔管中。电穿孔参数根据Fujii等人的方法(64)设置。
电穿孔后,立即将400微升预热的培养基(含5微摩尔CHIR99021)加入电穿孔管中。在37摄氏度下孵育40分钟后接种细胞。电穿孔两天后,通过荧光显微镜测量转染效率。电穿孔三天后,基于Nutlin-3a能抑制TP53野生型细胞增殖这一原理,用10微摩尔Nutlin-3a处理类器官2至3周,以实现对TP53突变细胞的功能性筛选。以电穿孔负对照RNP复合物的类器官作为对照组。
为验证靶向突变,从编辑后的类器官中提取基因组DNA,随后进行PCR扩增、基于拓扑异构酶的克隆以及Sanger测序。本部分所用的所有试剂和单导向RNA序列见表S2。
胃食管交界处类器官脂质组的基质辅助激光解吸电离成像质谱分析
将类器官从基质胶中分离出来后,用细胞回收液处理,并用冷PBS冲洗三次,然后转移到均匀的模具中,浸入M-1嵌入基质中。将类器官模具用铝箔包裹,漂浮在液氮上进行逐步冷冻。将冷冻的类器官平衡至-20°C,在Leica CM1860 UV冷冻切片机上以10微米的厚度进行冷冻切片,并将切片贴附在温度平衡过的、用己烷和乙醇清洗过的铟锡氧化物载玻片(Delta Technologies)上。
所有类器官均以能最大限度增加每张载玻片切片数量的布局进行切片,以便将TP53/CDKN2A KO与对照胃食管交界处类器官进行比较。为技术重复,以相同的布局对几个连续切片进行冷冻切片。
用HTX M5喷雾器将载玻片喷上DHB(40毫克/毫升),并用70%高效液相色谱(HPLC)级甲醇/30% HPLC级水溶解,参数如下:喷嘴温度-75°C,八次喷雾,流速0.1毫升/分钟,喷嘴速度1200毫米/分钟,3毫米轨道间距,交叉图案,压力10磅/平方英寸,气体流速3升/分钟,干燥时间10秒。最终的矩阵密度为8.89×10⁻¹毫克/平方毫米,线性流速为8.33×10⁻³毫升/毫米。
在约翰斯·霍普金斯应用成像质谱核心实验室的Bruker MALDI–time-of-flight (TOF)/TOF rapifleX 仪器(布鲁克道尔顿)上以反射正离子模式获取 MALDI-IMS 数据,像素大小为20微米,光栅为20微米,成像激光为20微米,每个像素200次激光照射,质量范围为 m/z 40 至 2000。使用 flexImaging 5.0 版和 flexControl 4.2 版(布鲁克道尔顿)收集成像数据,并将所有数据分析归一化到总离子流(TIC)。将 MALDI 脂质成像数据导入 SCiLS Lab 软件(2020a 版,SCiLS 有限公司),并使用 TIC 进行对齐以消除成像中的任何漂移伪影。在 TP53/CDKN2A KO 和对照 GEJ 类器官之间进行定量光谱、基于像素的配对比较(65-67)。
为了进行结构鉴定,使用氩气碰撞诱导解离的组织上MS/MS对顶部脂质的 m/z 进行鉴定,使用单束激光,结果区域为 54 微米 × 54 微米,4000 次激光照射,隔离窗口为 ±2 Da。从两种基因敲除类器官中收集 MS/MS 光谱。在初步鉴定时,通过将峰列表上传至脂质地图结构数据库(68),以±0.2 m/z的质量容差搜索 [M - H] 和 [M - Na],并选择所有脂质类别,从而生成潜在匹配物列表。这些潜在匹配物用于在 ChemDraw Professional 16.0 版(珀金埃尔默公司)中解析 MS/MS 光谱。
全基因组 DNA 甲基化谱分析及数据处理
使用 Illumina MethylationEPIC 阵列平台生成了四组对照和双基因敲除 GEJ 类器官的 DNA 甲基化图谱,该平台结合了基因组 DNA 的亚硫酸氢盐转化和全基因组扩增,以及直接基于阵列的 CpG 位点捕获和评分。使用 DNeasy 血液和组织试剂盒从类器官中提取基因组 DNA。所有 DNA 样本均通过 Qubit 双链 DNA 广泛范围(BR)测定法进行定量,通过 A260/280 和 A260/230 比率评估纯度,并通过在 0.8% 琼脂糖凝胶上电泳检查完整性。然后将 DNA 样本按照 Infinium HD 甲基化测定法协议(69)与 Infinium MethylationEPIC 芯片杂交。
使用 SeSAME 软件包(70)中的 OpenSesame 函数提取每个探针的 DNA 甲基化值。根据 Infinium DNA 甲基化阵列的注释文件移除推荐的一般屏蔽探针。通过 Limma 软件包(版本 3.46.0)以校正 P 值 < 0.05 且绝对甲基化变化量 > 0.2 的标准识别差异甲基化探针。研究者进一步利用 DMRcate 软件包基于差异甲基化探针识别差异甲基化区域(DMRs),采用 Fisher 确切概率检验 P 值 < 0.05 的标准。
RNA 测序及数据分析
对来自四名患者的四组对照和双基因敲除 GEJ 类器官的配对样本进行了 RNA-seq 分析。提取总 RNA 并用脱氧核糖核酸酶 I(DNase I)处理后进行测序。使用 NEBNext Ultra Directional RNA Library Prep 试剂盒构建文库。定量后的文库在 Illumina NovaSeq 6000 平台上进行测序,并生成了配对读段。构建参考基因组索引,使用 HISAT2 v2.0.5 将配对的干净读段与参考基因组进行比对。通过 FeatureCounts v1.5.0-p3 生成每个基因的读段计数。根据基因长度和映射到该基因的读段计数计算每个基因每百万映射片段的外显子每千碱基片段数。使用 DESeq2 结果进行差异表达分析,将 DESeq2 找到的校正 P 值 < 0.05 且绝对倍数变化 > 2 的基因指定为差异表达基因。使用 clusterProfiler R 包(71)对差异表达基因进行 GO 富集分析。
实时定量聚合酶链式反应
使用 RNeasy 试剂盒提取总 RNA,并通过柱上 DNase 消化去除基因组 DNA。使用 iScript Select cDNA 合成试剂盒(Bio-Rad)对总计 500 μg 的 RNA 进行反转录。定量 PCR 使用 iTaq Universal SYBR Green Supermix 进行。所有结果均以 β-肌动蛋白(β-actin)表达为内参进行标准化。用于定量逆转录(qRT-PCR)的引物信息见表 S2。
小干扰 RNA(siRNA)介导的基因沉默
电穿孔前两天,将类器官传代培养在不含抗生素的类器官培养基中,培养基中添加 5 μM CHIR99021。将类器官解离为由 10 至 15 个细胞组成的团块,并用 100 μl 含 4 μM 电穿孔增强剂的电穿孔缓冲液重悬,随后加入 10 μl 50 mM 的 siRNA 溶液。电穿孔 24 小时后,重复上述操作再次进行电穿孔。之后从形成的克隆类器官中提取总 RNA,反转录为 cDNA,并通过 qRT-PCR 检测 siRNA 的敲低效率。siRNA 序列详见表 S2。
组织学、免疫荧光(IF)和免疫组化(IHC)
类器官培养物和组织在室温下用 10% 福尔马林固定过夜。将石蜡包埋的类器官和组织连续切片,切片厚度为 10 微米。石蜡切片经脱蜡和复水处理后,进行苏木精-伊红(H&E)染色,或在接近沸腾的 10 毫摩尔柠檬酸钠缓冲液(pH 6.0)中进行抗原修复 10 分钟。
在进行免疫荧光(IF)染色时,载玻片首先在含 0.5% Triton X-100 的 PBS 中透化,再在含 1% 山羊血清的 PBS 中于室温封闭 30 分钟。封闭后,将载玻片置于湿润培养箱中,4°C 下与抗 Ki67(1:200,Abcam)或抗 TFF3(1:100,Abcam)抗体孵育过夜。切片随后用含 0.1% 吐温 20 的 PBS(PBST)洗涤(三次,每次 5 分钟),然后与 Alexa Fluor 标记的二抗(1:500)孵育 1 小时。再次用 PBST 洗涤后,使用含 4',6-二脒基-2-苯基吲哚(Sigma-Aldrich)的 Fluoroshield 封片。
免疫组化(IHC)染色使用徕卡 BOND-MAX 自动染色仪进行,所用抗体为细胞角蛋白 AE1/AE3(1:200,Santa Cruz Biotechnology)。图像采集由约翰斯·霍普金斯医学罗斯影像中心使用徕卡倒置共聚焦显微镜 SP8 完成。本部分使用的所有试剂详见表 S2。
染色质免疫沉淀法
染色质免疫沉淀(ChIP)实验使用 EZ-Magna ChIP A/G ChIP 试剂盒(默克密理博,17-10086)按照制造商提供的操作流程进行。细胞先用 1% 甲醛进行交联处理,随后使用含有 1× 蛋白酶抑制剂混合物 II 的细胞裂解液进行裂解。核组分用补充了 1× 蛋白酶抑制剂混合物 II 的核裂解液分离。
提取得到的染色质在冰上超声处理,总时长为 8 分钟,设置为 30% 振幅,脉冲开启 10 秒、关闭 20 秒,使其剪切为 200 至 1000 个碱基对的片段。剪切后的交联染色质与磁性蛋白 A/G 珠子及抗体共孵育以完成免疫沉淀。使用的抗体包括抗 FOXM1(每次 ChIP 反应加入 5 微克,Invitrogen,702664)和正常小鼠免疫球蛋白 G(IgG,1 微克/反应)作为阴性对照。
免疫沉淀后提取纯化 DNA,并通过定量 PCR(qPCR)进行检测。所用的 qPCR 引物详见表 S2。
裸鼠的异种移植
所有涉及小鼠的实验程序与方案均经约翰斯·霍普金斯大学医学院动物实验委员会批准。对于异种移植实验,将2×10⁶个类器官细胞或Eso26细胞重悬于冷却的50%基质胶中后注射至裸鼠腋窝处。
在WEB2086治疗实验中,实验小鼠每两天接受一次腹腔注射,剂量为5毫克/千克/天,持续3周。对照组则接受溶剂对照注射(即1.25%二甲基亚砜溶于磷酸盐缓冲液中)。异种移植物的尺寸每周测量两次,使用游标卡尺记录长度(L)与宽度(W),并根据公式V = 1/2 × (L × W²)计算异种移植物体积(V)以监测其生长情况。
实验结束后,对小鼠实施安乐死,并取出异种移植物用于后续分析。
统计分析
数据以平均值±标准差表示,除非另有说明。统计分析使用GraphPad Prism 9.2软件进行。对于体外实验,除非图例中另有说明,均采用t检验或单因素方差分析(ANOVA)。对于异种移植实验,采用非配对t检验。P < 0.05 被认为具有统计学意义。

汇报人:王肖宇
导师:赵宇
审核:邱轲、任建君
