


原创 胥飞宇 华西医院耳鼻喉科
华西耳鼻喉学术前沿速递——文献精读(第69期)
精读分享│【Cancer Discovery】:HPV整合诱导的瘤内异质性和克隆进化
英文题目:Intratumoral Heterogeneity and Clonal Evolution Induced by HPV Integration
中文题目:HPV整合诱导的瘤内异质性和克隆进化
期刊:Cancer Discovery(IF: 30.6)
单位:德克萨斯大学安德森癌症中心胸/头颈肿瘤科
发表时间:2024年9月


摘要:
人乳头瘤病毒(HPV)基因组在大多数HPV阳性癌症中会整合到宿主DNA中,但其对染色体完整性的影响尚不清楚。通过对口咽癌和癌细胞系进行连续长读长测序,研究者发现了一种此前未被描述的结构变异(SV)形式——“异质链”(heterocateny)。其特征为肿瘤内部存在多样化、相互关联且重复的病毒和宿主DNA片段的串联结构。SV之间共享的独特断点有助于逐步重建它们从一个共同分子祖先的演化过程。该分析表明,病毒及病毒-宿主串联体不稳定,在插入和从染色体中切除时,会促进宿主DNA的捕获、扩增和重组,并引发染色体重排。该研究在染色体外DNA(ecDNA)和染色体DNA(icDNA)中均检测到异质链的存在。这些发现表明,异质链是由致癌DNA病毒的动态异常复制和重组驱动的,从而扩展了HPV整合的已知后果,包括促进肿瘤内异质性和克隆进化。
意义:通过对HPV阳性癌症的长读长测序,揭示了“异质链”这一以前未报道的基因组SV形式,其特征是肿瘤内部存在异质性、相互关联且重复的基因组重排。异质链由不稳定的HPV基因组串联体驱动,促进了宿主DNA的捕获、重排和扩增,并推动了肿瘤内异质性和克隆进化。
思维导图:
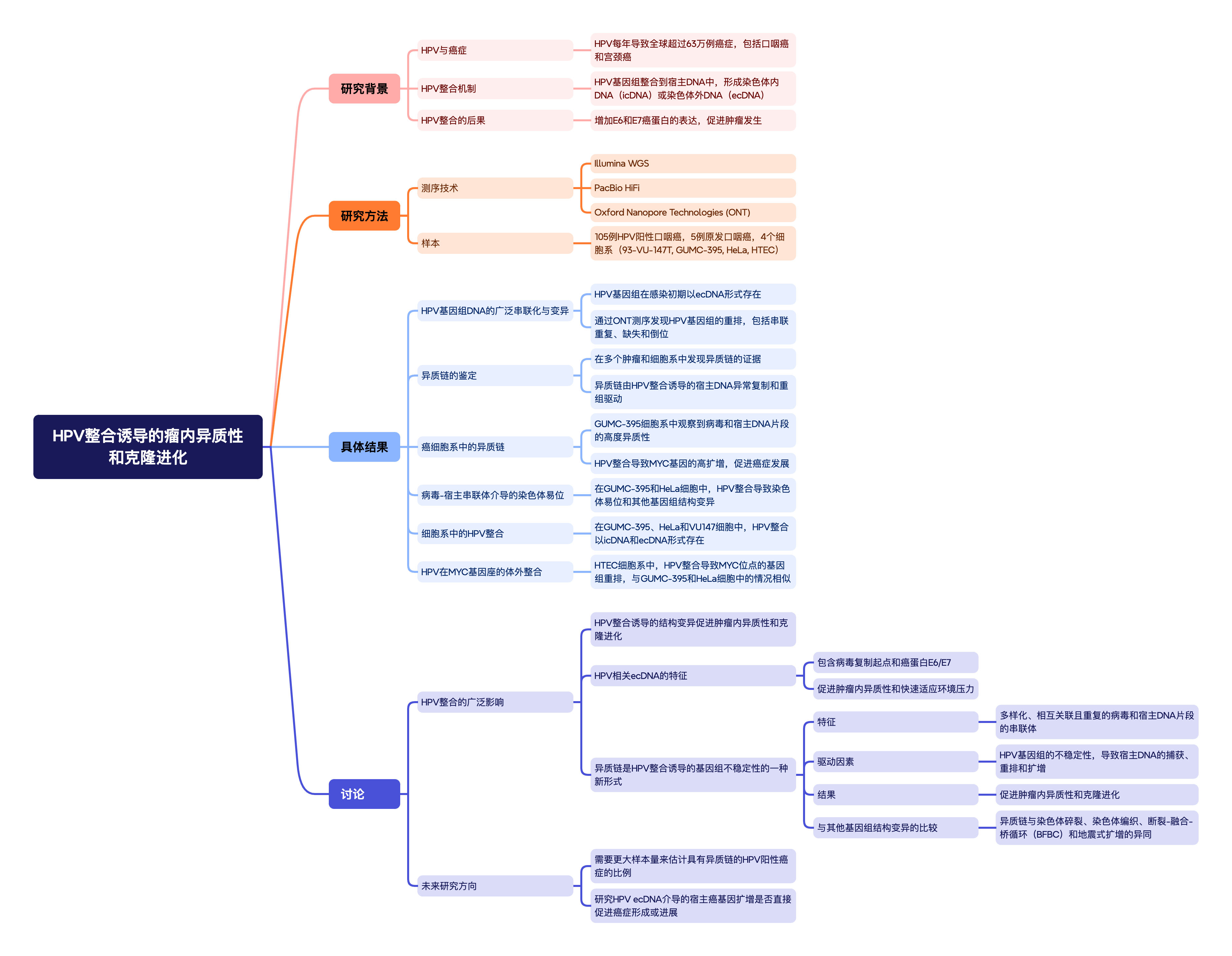
研究背景:
人乳头瘤病毒(HPV)每年在全球导致超过63万例癌症,包括肛门生殖器和口咽鳞状细胞癌。在感染早期,病毒基因组以约8千碱基对(kb)的染色体外环状DNA(ecDNA)形式存在,即游离体(episome)。在大多数后续产生的癌症中,HPV DNA已整合到宿主基因组中,通过断点将病毒和细胞DNA连接起来,形成染色体内DNA(icDNA)和/或ecDNA形式。HPV整合通过增加编码E6和E7癌蛋白的转录本表达和稳定性,促进肿瘤发生。其中,E6靶向降解肿瘤抑制蛋白p53,E7则靶向降解pRb。最近对宫颈癌和口咽癌的全基因组测序(WGS)分析显示,HPV整合位点富集于具有结构变异(SV)和拷贝数变异(CNV)的基因组区域。现已发现了HPV整合的多种遗传后果,包括整合位点附近宿主基因表达的失调。
要更好地理解HPV整合如何导致SV、CNV以及宿主基因表达异常,需要提高基因组序列变异及其连接性的分辨率。为了解析HPV整合位点两侧的基因组重排结构,研究者对HPV阳性口咽癌和人类细胞系进行了连续长读长测序(LR-seq)。该分析揭示了一种基因组SV形式,研究者将其命名为“异质链”(heterocateny,意为“可变链”)。异质链的特征是肿瘤内部存在多样化、相互关联且重复的病毒和宿主DNA片段的串联体。基于长读长测序数据的进化模型将异质链解释为HPV整合诱导的宿主DNA异常复制和重组的结果。研究者得出结论,HPV整合促进了肿瘤内异质性和克隆进化。
主要结果:
研究者对105例HPV阳性口咽癌进行了WGS,发现HPV-宿主断点直接位于富含SV和CNV的宿主基因组区域两侧、桥接或映射其中。为了解析HPV整合位点的基因组重排,研究者在此使用了Illumina WGS以及两种长读长测序技术:PacBio HiFi和Oxford Nanopore Technologies(ONT)。选择这些方法是因为它们能够生成单核苷酸水平上错误极少的高分辨率读长(WGS、PacBio),或能够跨越包括重复元件在内的基因组特征、长度可达数十千碱基的连续长读长(ONT)。根据ONT读长的预期长度,研究者选择了5例HPV阳性原发口咽癌和4个细胞系,每个样本的病毒-宿主断点均通过WGS定位到拷贝数(CN)≥4n的CNV靶区域和/或断点间隔<60 kb的SV。在通过WGS研究的105例口咽癌肿瘤中,45%的整合病毒肿瘤在HPV插入断点1兆碱基对(Mbp)范围内观察到CN≥4n的CNV。细胞系包括93-VU-147T、GUMC-395、HeLa和HTEC。
1.HPV基因组DNA的广泛串联化与变异
在初始感染阶段,HPV以约7.9 kb的ecDNA游离体形式存在于细胞核中。因此,研究者评估了两种长读长测序方法捕获和识别小环状DNA分子的技术能力,以内源性约16.5 kb的环状线粒体DNA(mtDNA)基因组作为参考。ONT测序的mtDNA读长直方图显示频率峰值出现在16.5kb和33kb处(Figure S2.1)。映射读长的5′和3′端之间的距离在参考mtDNA基因组中小于100碱基对(bp),表明主要存在单体和双体环状mtDNA基因组。这一分析证实了LR-seq能够检测ecDNA、确定其长度并识别头尾串联重复序列。
对映射到HPV参考基因组的ONT读长的类似分析显示,读长经常超过约7.9 kb(Figure 1A–D;Figure S2.2)。映射读长的5′和3′端之间距离的图(Figure 1E)表明,在一例原发癌症中主要为单体HPV游离体(Figure 1A,tumor 1),而在其他样本中则存在多头尾病毒-病毒串联体(Figure 1A–D,Figure 1F;Figure S2.2),这与最近的报道一致。
与mtDNA相比,HPV基因组的ONT读长更频繁地偏离预期的单位长度(即约7.9 kb的倍数;Figure 1A–D),揭示了病毒DNA中的重排。所有独特的病毒-病毒断点均通过至少两种测序平台(通常覆盖三种技术)得到确认,排除了技术假象的可能性。将VU147、肿瘤2和肿瘤3的ONT读长与HPV16模板模型比对,揭示了包括串联重复、缺失和倒位在内的重排(Figure 1G;Figure S2.3)。在VU147中检测到的七个独特病毒-病毒断点分别被赋予数字标识(即1–7)。为了便于模式识别,使用块状图可视化LR-seq读长中的DNA片段,并使用断点图可视化断点(Figure 1H)。在这些及后续的所有可视化图中,HPV参考基因组坐标0用黑色或红色垂直线表示(Figure 1–6)。VU147中HPV DNA的多种多样重排显而易见(Figure 1H),表明串联体病毒基因组的遗传不稳定性。
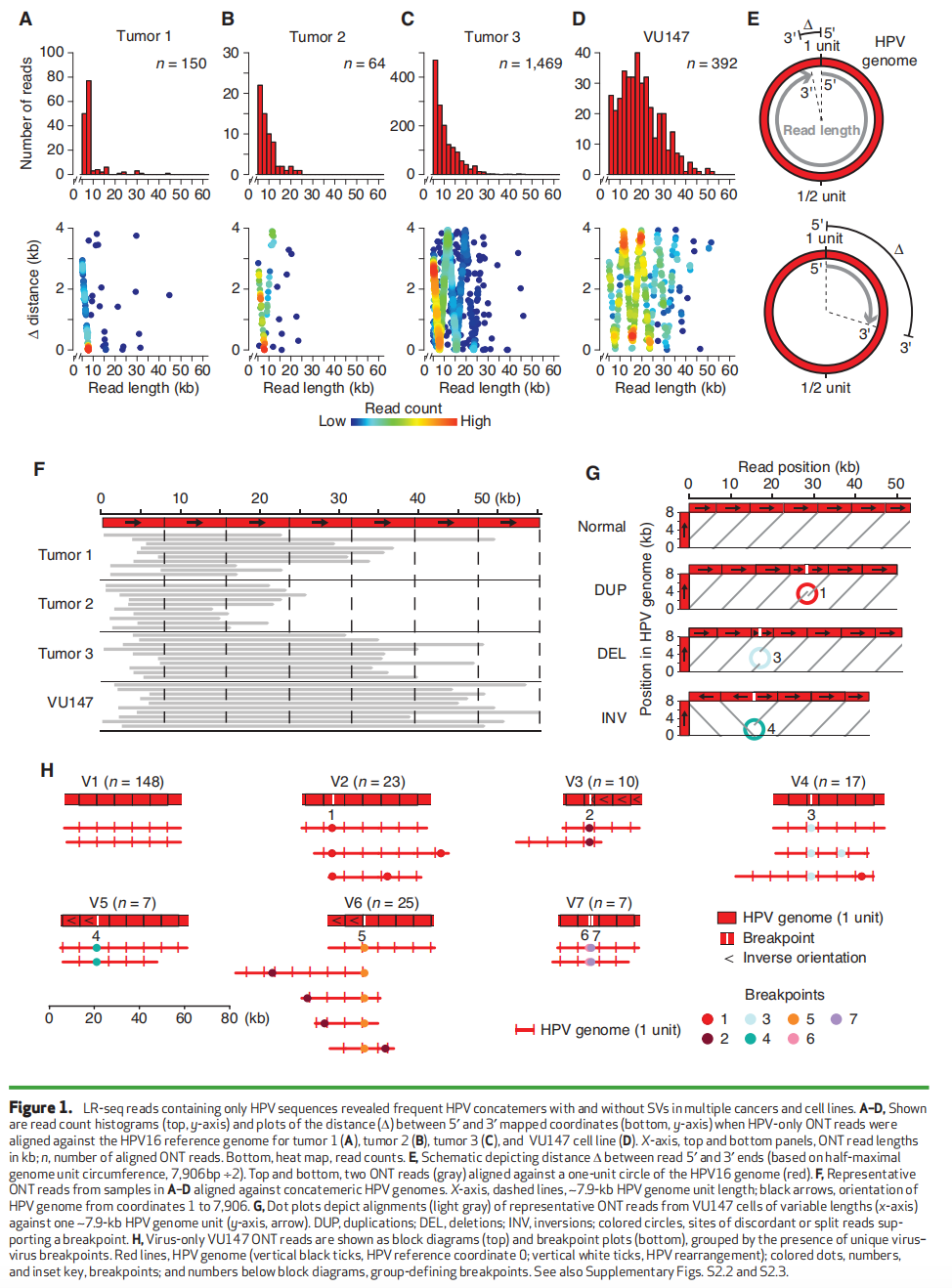
Figure 1. LR-seq reads containing only HPV sequences revealed frequent HPV concatemers with and without SVs in multiple cancers and cell lines.
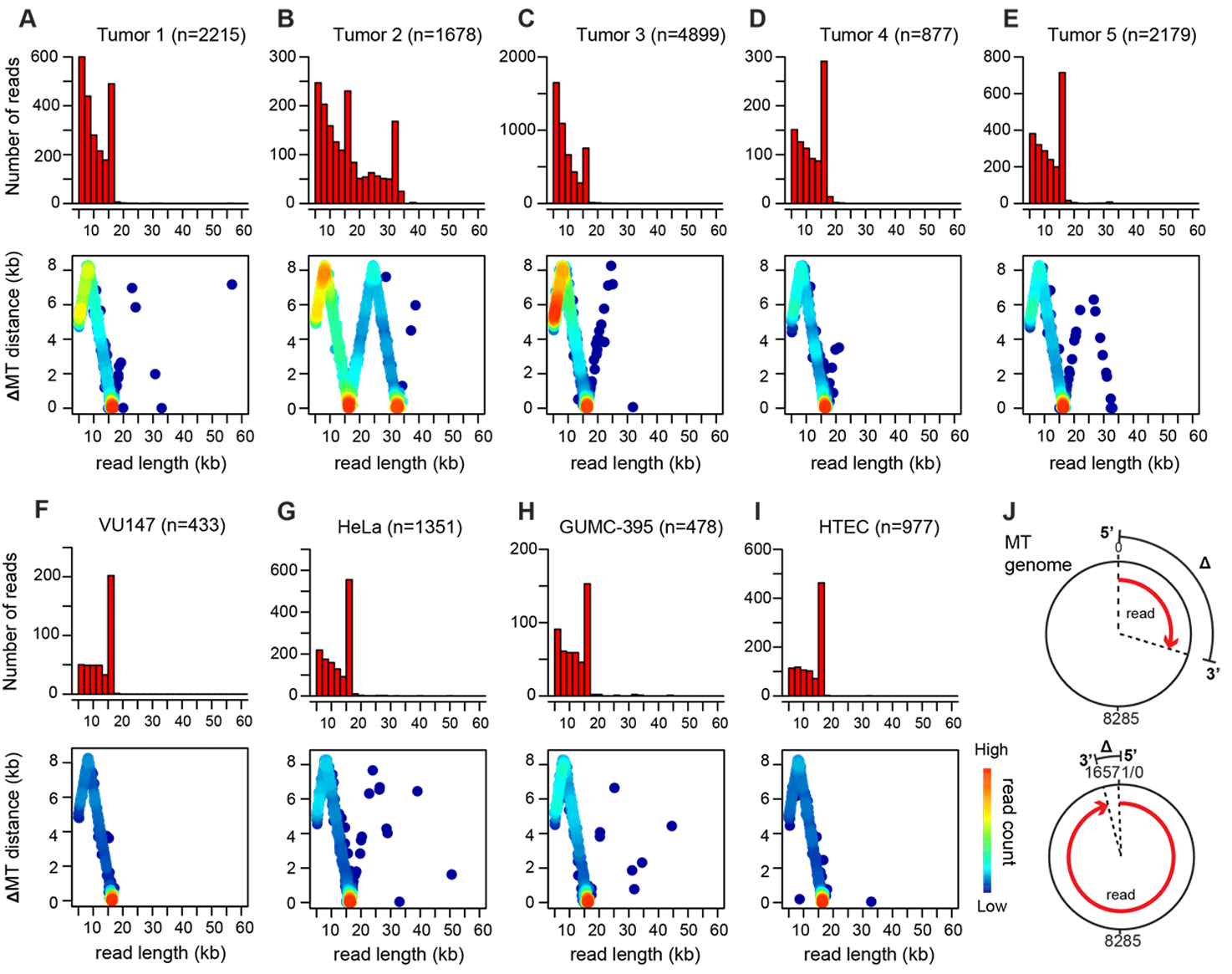
Figure S2.1. Detection of circularized mitochondrial DNA by LR-seq reads.
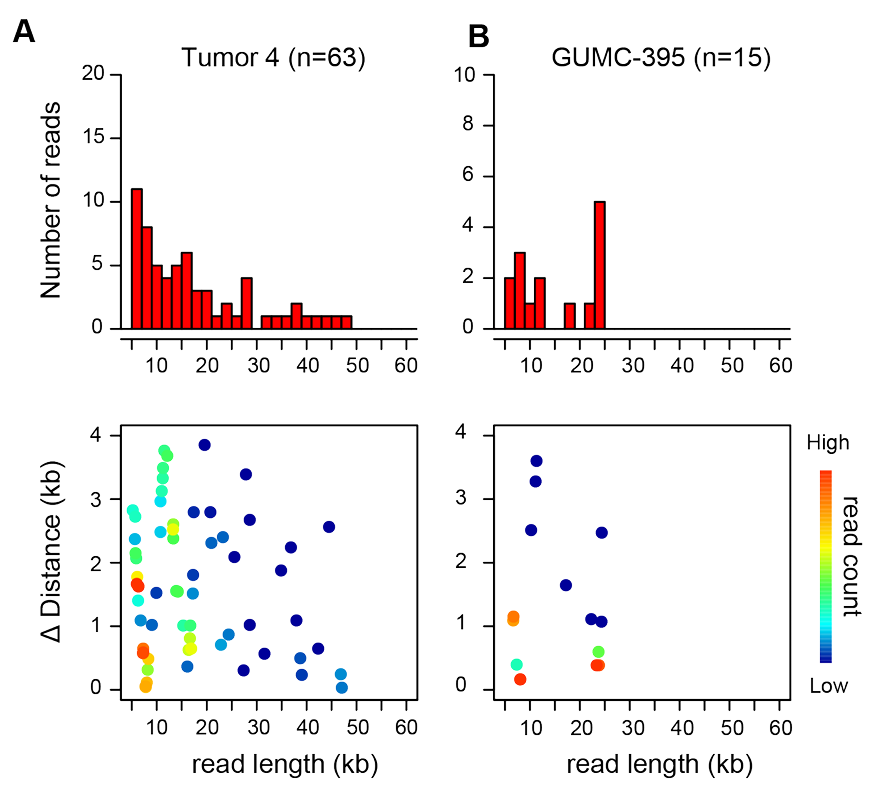
Figure S2.2. Detection of HPV concatemers by LR-seq reads.
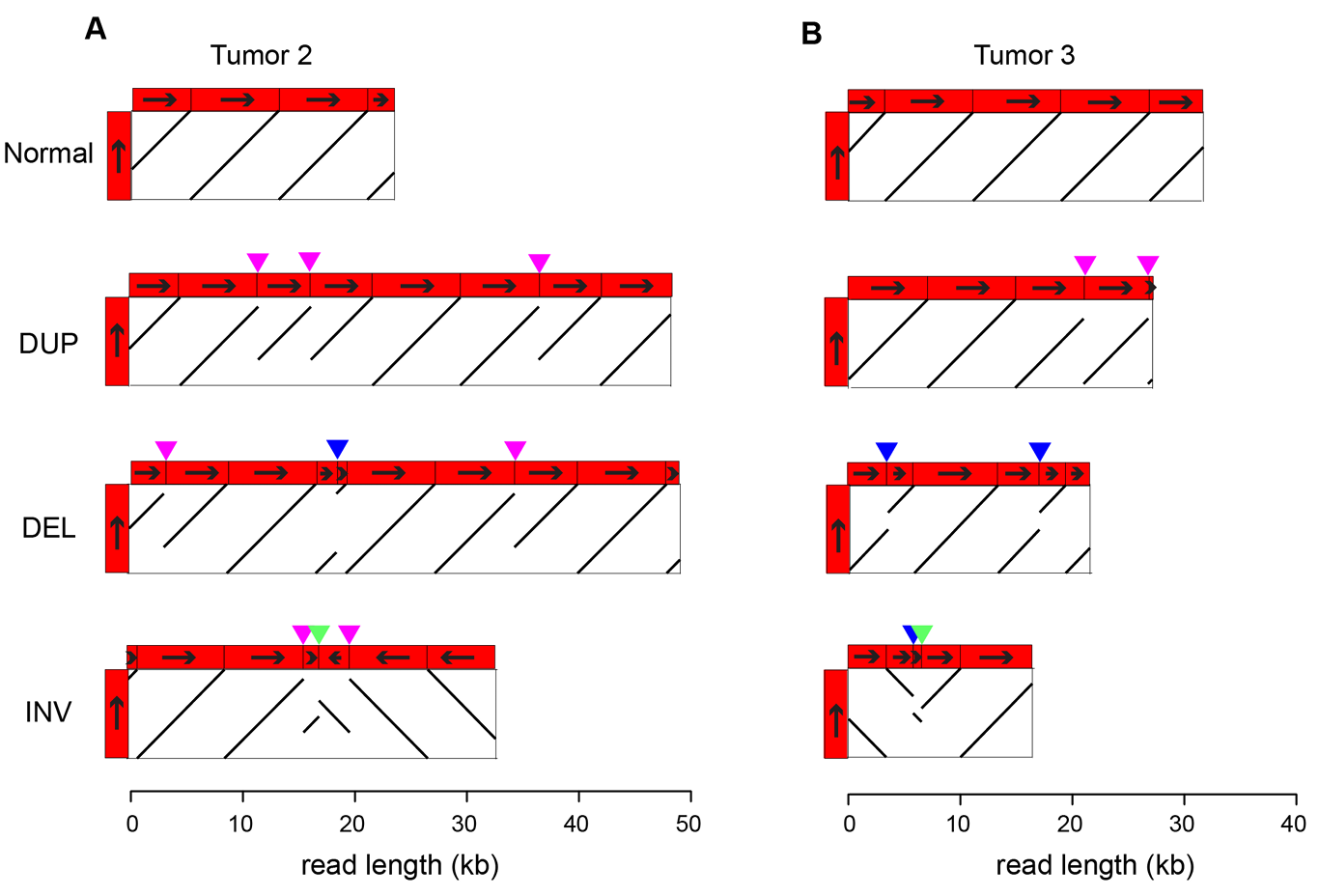
Figure S2.3. ONT reads show structural rearrangements in HPV concatemers.
2.异质链的鉴定:一种独特的结构变异形式
通过对病毒-宿主断点两侧的基因组SV进行扩展分析,研究者发现肿瘤4在5p13、5q14和Xp22染色体上共有22个独特的断点,这些断点位于CNV和SV区域的两侧,其中包括14个HPV-宿主断点、5个宿主-宿主断点和3个病毒-病毒断点(Figure 2A)。重排包括两个染色体易位:t(5;X)(p13;p22)和t(5;X)(q14;p22)。为了解析ONT读长覆盖的基因组结构重排,研究者选择了由不一致或分裂的WGS和/或LR-seq读长支持的最佳断点作为片段定义断点。这使得研究者能够基于参考人类基因组和HPV类型特异性基因组来划分宿主或病毒DNA片段。
根据WGS估计,肿瘤4中每个单倍体基因组含有约171个HPV16基因组拷贝。检测到由多达六个串联HPV16基因组组成的病毒-病毒串联体(Figure 2B,group A1),但在相邻病毒基因组单元中间歇性删除了HPV核苷酸5,144至7,906以及1至776,形成了一个独特的、反复出现的病毒-病毒断点(即断点20;Figure 2B,group A2)。长度≥20 kb的ONT读长(N = 178)揭示了重排的病毒-宿主结构,其中X染色体(如XB和XD)和/或5染色体(如5B、5E和5G)的特定片段精确插入到病毒基因组片段被删除的位置(Figure 2B,group A3–10)。单个分子显示出特定的病毒和/或宿主DNA片段及断点模式,在某些情况下这些模式会重复出现(Figure 2B,group A6、9、10)。这些多样化的模式与VU147中仅包含病毒的ONT读长观察到的模式类似(Figure 1H)。研究者根据关键断点将肿瘤4的读长聚类为不同组(Figure 2B,group A1–10)。在这些读长组内部和组之间,断点模式显著不同,显示出广泛的分子间异质性。定义某一组的独特断点和片段模式偶尔会与其他组的模式在单个分子中连接。例如,组A3中的断点13也与组A5中的断点12和14相连(Figure 2B)。
研究者利用肿瘤4中异质结构共享的独特断点和模式作为分子条形码,从共同的分子祖先重建基因组结构演化过程。根据生成的机制模型,串联化的HPV基因组最初插入到X染色体上宿主DNA片段XC的缺失位点(Figure 3)。随后,这些串联化的HPV基因组从X染色体切除时捕获了宿主DNA并形成ecDNA,然后插入到5p和5q染色体中(Figure 3)。共享的病毒和宿主DNA片段及断点以重复模式串联连接,但缺乏单核苷酸变异(SNV)或小的插入/缺失(indel;Figure S3.1),这与通过同源重组和间歇性高保真滚环复制或依赖重组的复制(RDR)形成的机制一致。
在肿瘤4中,支持重排的病毒-宿主串联体整合到侧翼染色体DNA的读长数量非常少。因此,研究者推断在该肿瘤中观察到的众多病毒-宿主串联体主要以ecDNA形式存在。值得注意的是,与LR-seq检测到的病毒-宿主串联体相比,AmpliconArchitect软件对ecDNA的预测过于简化且不准确(Figure 2B;Figure S3.2),这可能是由于短读长WGS数据的固有局限性。
总之,肿瘤4的LR-seq数据揭示了病毒-宿主断点两侧基因组SV的显著程度,其特征是多样化、相互关联且重复的病毒和宿主DNA片段及断点模式。研究者将这种基因组SV形式命名为“异质链”。在所有研究的癌症和细胞系中均观察到了异质链的多个独立证据,如下所述。
肿瘤2在22q13.2染色体上的约60 kb EP300位点共有23个断点,包括14个病毒-宿主断点、4个宿主-宿主断点和5个病毒-病毒断点。EP300在HPV阳性口咽癌中经常因体细胞突变而失活。其中14个断点被选为片段定义断点(Figure 4A)。与肿瘤4类似,研究者使用关键断点将ONT读长分组(Figure 4B)。ONT读长(N =154)支持包含多个串联全长HPV基因组单元的串联体,其中穿插着缺失核苷酸7,065至7,906和1至2,312的串联体(断点17;Figure 4B,group B2)。包含断点17的病毒串联体被检测到与EP300片段串联(Figure 4B,group B3–10)。读长组内部和组之间的结构异质性,类似于肿瘤4中的情况(Figure 2B),进一步证明了异质链的存在。根据LR-seq数据建立的模型,这些结构从克隆祖先通过一系列事件演化而来,包括串联化HPV基因组的插入、ecDNA切除、拷贝数扩增以及其他重排(如连续缺失;Figure S4.1)。没有WGS或LR-seq读长支持病毒-宿主结构整合到22q13.2染色体中,表明包含EP300片段的病毒-宿主串联体主要或仅以ecDNA形式存在。
有趣的是,肿瘤2中的ONT读长独立鉴定了病毒-宿主串联体整合到4p15.31染色体侧翼宿主序列的情况(Figure 4C)。在4染色体(Figure 4D)和22染色体EP300位点检测到相同的断点17,表明这两个不同位点的病毒-宿主串联体具有克隆相关性。这一例子表明,串联体可以同时以ecDNA和icDNA整合形式存在于同一肿瘤中,并且相同的断点可以在两种基因组DNA形式中找到。
在肿瘤5中检测到的病毒-宿主串联体映射到8q24.21染色体上的癌症驱动基因MYC附近或内部,这是口咽癌和宫颈癌中HPV整合的热点区域。研究者鉴定了6个断点,包括3个病毒-宿主断点和3个宿主-宿主断点,并选择这些断点来划分MYC位点的宿主片段A至J(Figure S4.2A)。尽管未检测到HPV串联体,但鉴定出HPV核苷酸1,803至2,170的缺失。研究者检测到110条ONT读长(每条≥20 kb)定义了MYC位点的SV。其中98条(88%)支持MYC至少串联重复两次的基因组重排(片段E,Figure S4.2B)。较少见但相关的SV通过重组事件从这一祖先分子衍生而来。由于没有读长支持病毒-宿主串联体整合到相邻染色体DNA中,它们可能主要以ecDNA形式存在。由于该肿瘤每个单倍体基因组含有约20个HPV16基因组拷贝,每个细胞可能包含一系列ecDNA分子,其长度与连接的HPV单元数量相称(Figure S4.2C)。与肿瘤4和2相比,肿瘤5中ecDNA结构的相对同质性表明,捕获的MYC癌基因赋予了选择性克隆生长优势。
在肿瘤3中,以1至6个基因组单元为主的HPV16游离体占主导地位(Figure 1C和F)。5个病毒-宿主断点映射到3q27.1染色体上的基因丰富区域(Figure S4.2D),LR-seq数据支持病毒串联体插入该位点(Figure S4.2E-F)。相对较低的读长数量和较少的衍生重排(Figure S4.2E,group E3–5)表明,ecDNA切除和重组可能发生在亚克隆细胞群体中。因此,肿瘤5和3分别由携带HPV整合诱导的ecDNA的主要克隆或亚克隆细胞群体组成。
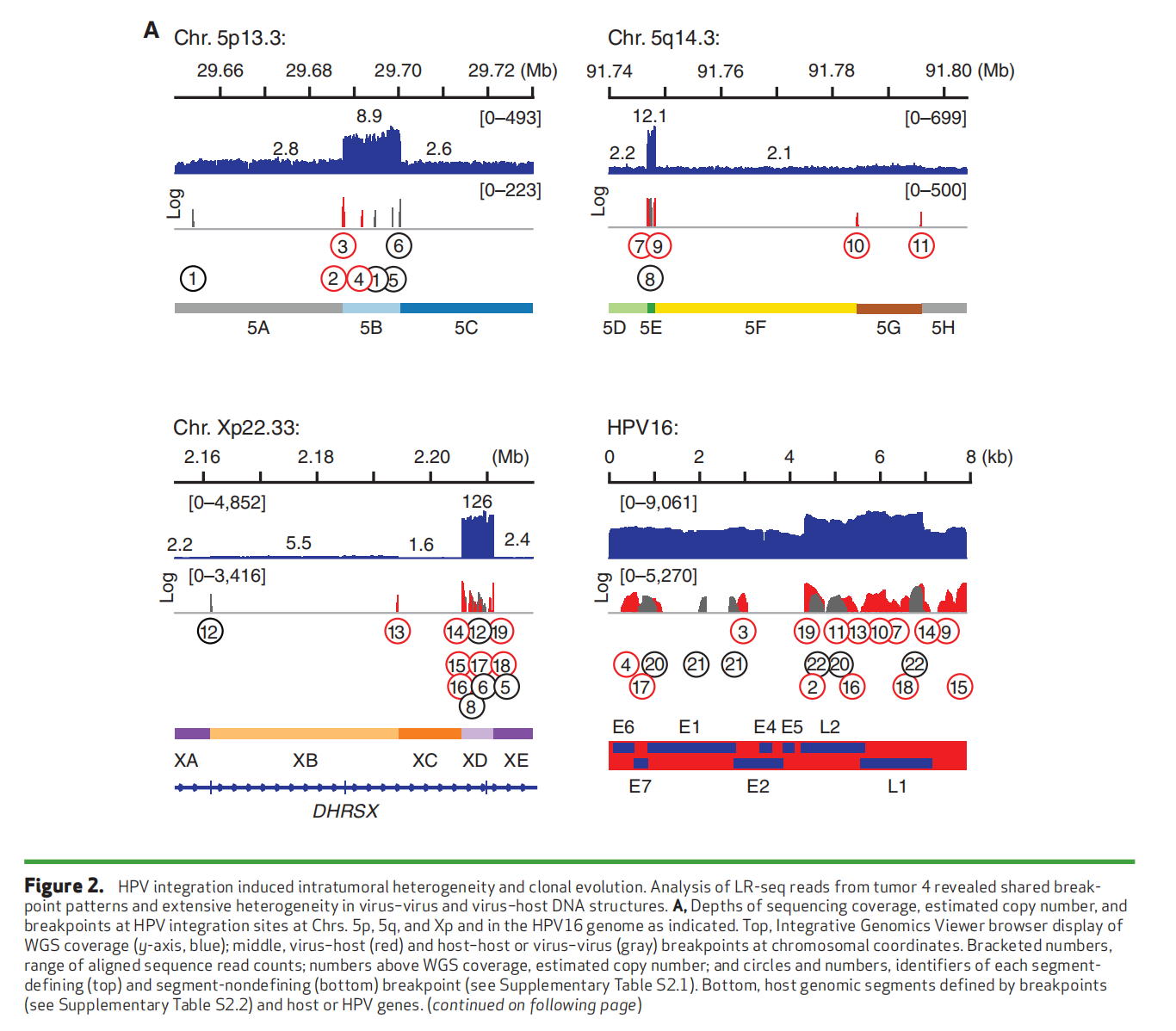
Figure 2. HPV integration induced intratumoral heterogeneity and clonal evolution.
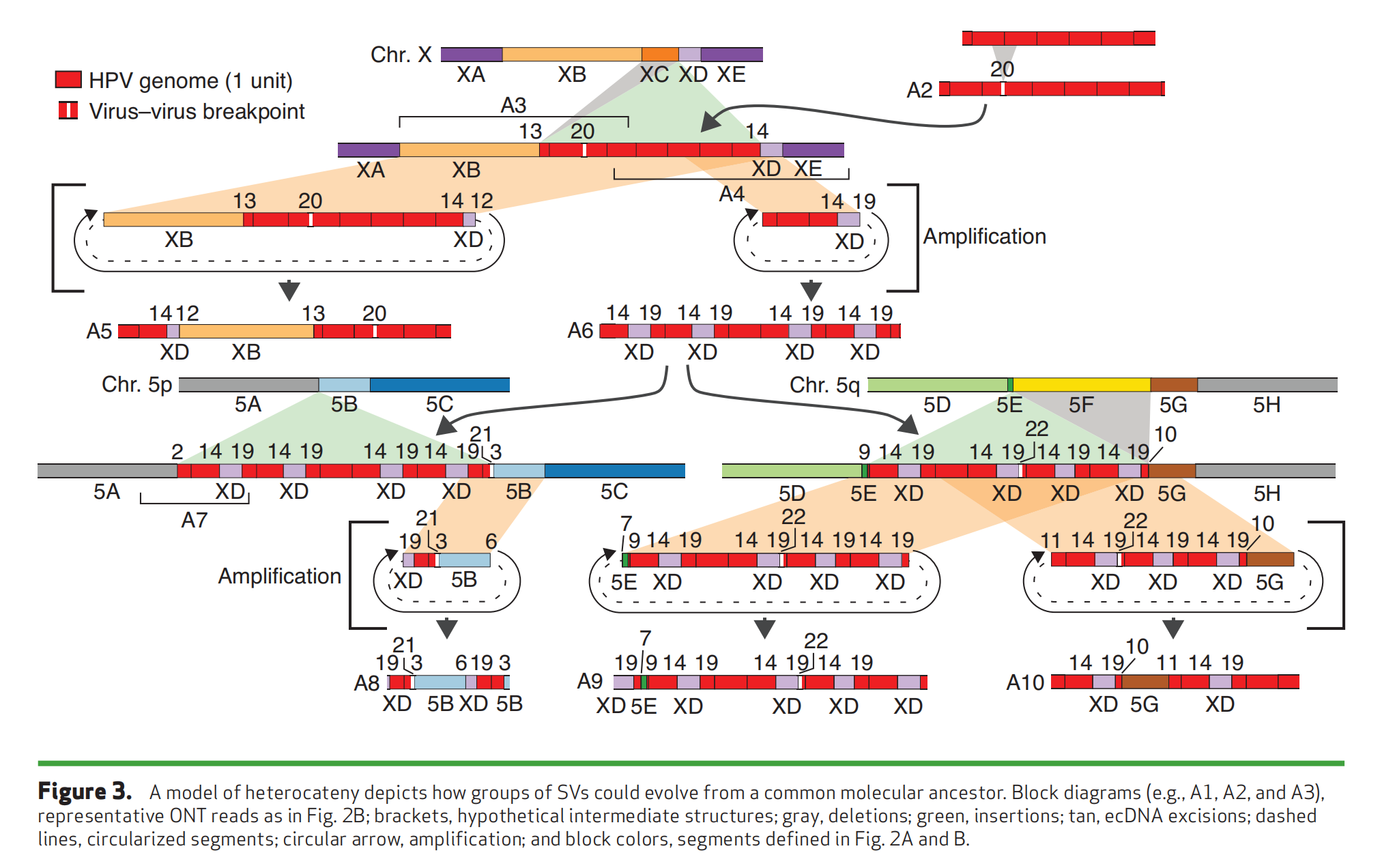
Figure 3. A model of heterocateny depicts how groups of SVs could evolve from a common molecular ancestor.
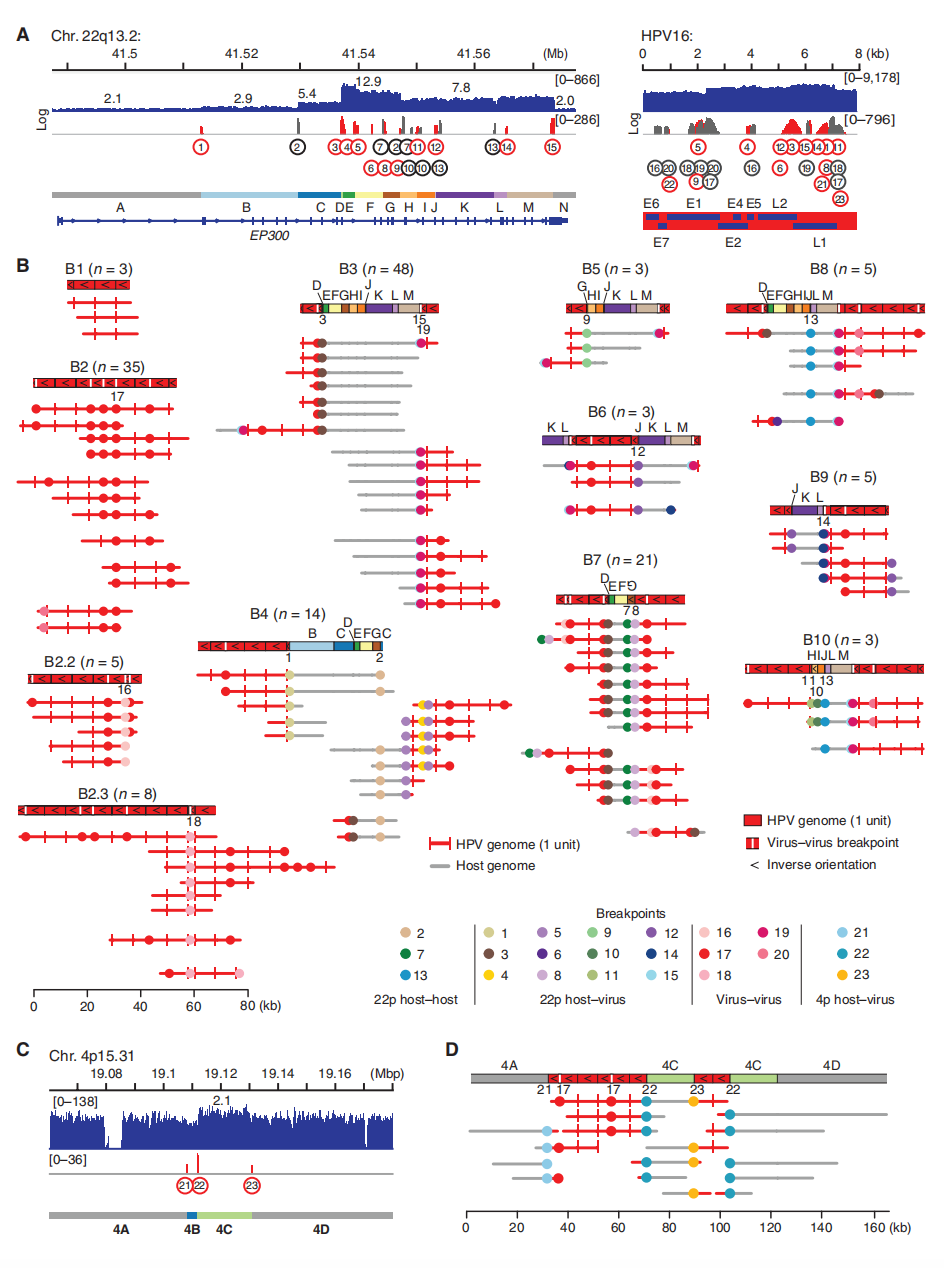
Figure 4. Heterocateny disrupted the EP300 locus and Chr. 4p15 in tumor 2.
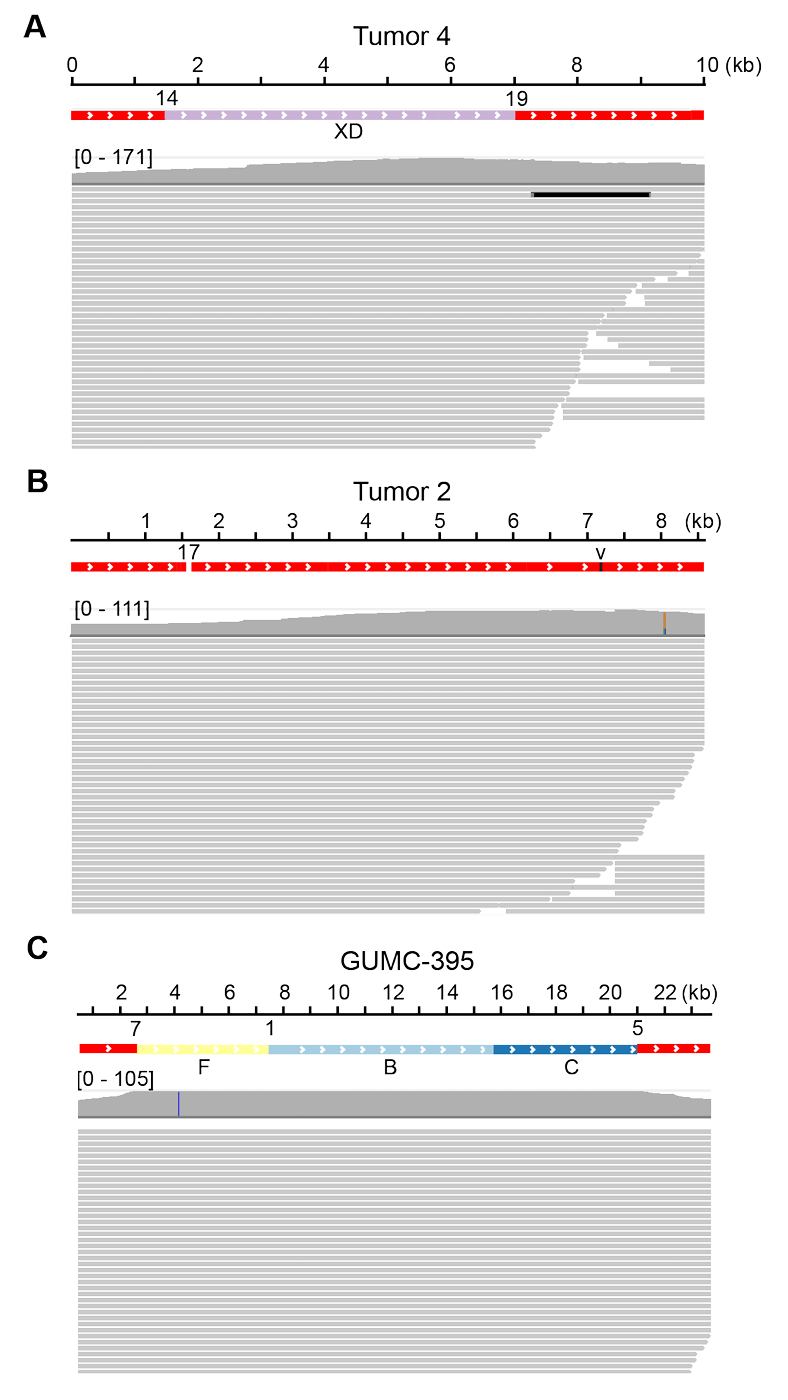
Figure S3.1. Absence of sequence variants at breakpoints.
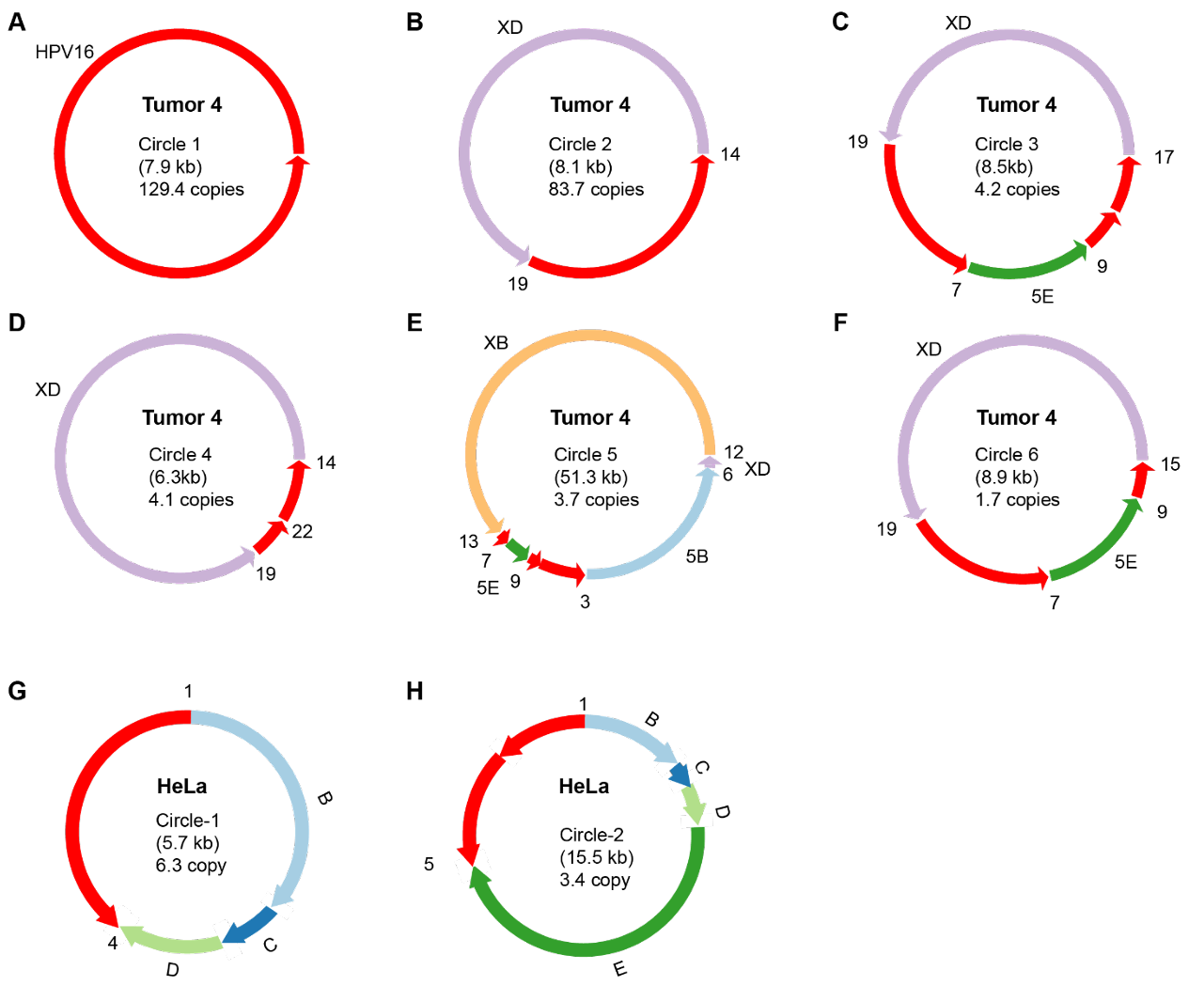
Figure S3.2. AmpliconArchitect predicts inaccurate HPV ecDNA structures.
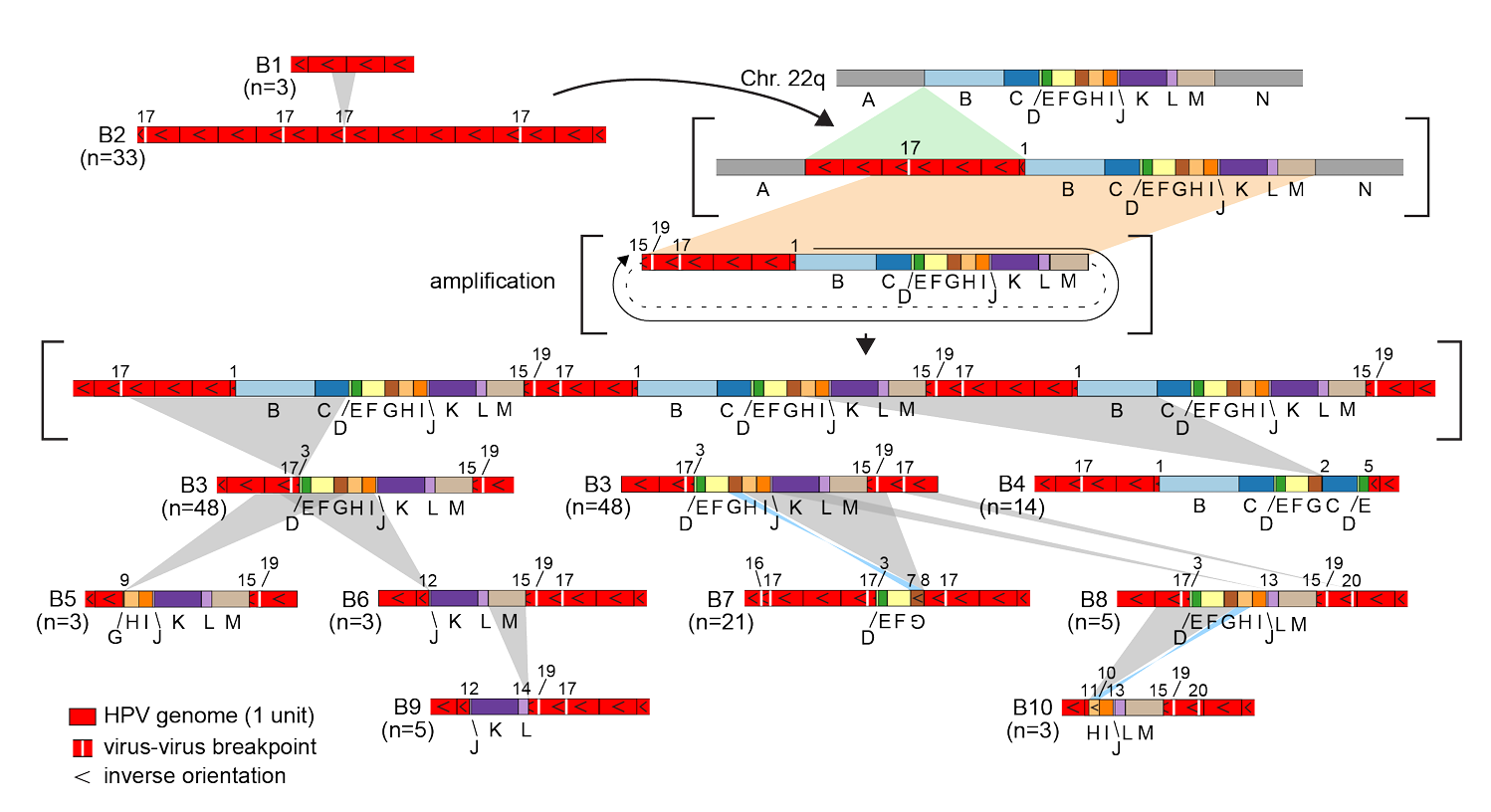
Figure S4.1. Evolution of structural variants at Chr. 22 of Tumor 2.
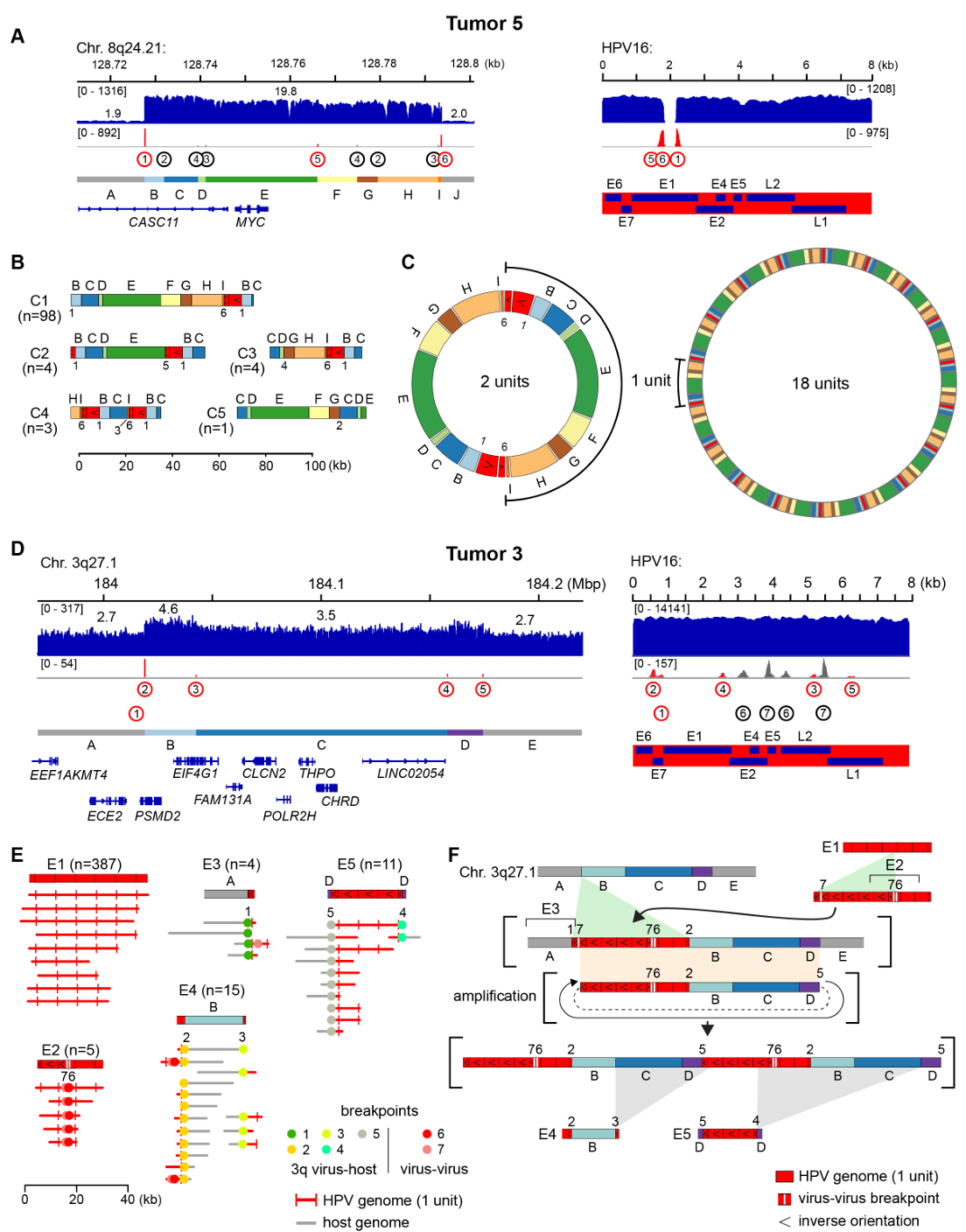
Figure S4.2. Evidence of heterocateny in Tumor 5 and Tumor 3.
3.癌细胞系中的异质链
GUMC-395细胞系来源于一种侵袭性宫颈神经内分泌癌的肝转移灶。GUMC-395细胞含有13个断点,包括5个病毒-宿主断点和7个宿主-宿主断点,这些断点集中在MYC位点附近约200 kb的极端高扩增区域(高达约225n)和结构重排区域(Figure 5A)。其中8个断点定义了连续的宿主DNA片段A至L。片段B和C包含MYC基因。该细胞系每个单倍体基因组含有约112个HPV拷贝。与研究者在原发癌症中的观察类似,GUMC-395细胞的ONT读长中观察到病毒和宿主DNA片段及断点模式的显著异质性(Figure 5B)。在8号染色体的片段E和F之间检测到病毒串联体的插入(Figure 5A),定义了断点6和7。有趣的是,没有序列数据支持连接宿主DNA片段E到F的正常等位基因,表明存在杂合性丢失。在ONT读长中鉴定的大多数病毒-宿主串联体(N = 774,≥20 kb)包含断点7,表明这一插入是一个早期的、可能是致瘤性事件。此外,许多SV共享相同的V–F–B–C模式,包含MYC基因以及宿主片段D和E的缺失(Figure 5B),这与从共同分子祖先演化的过程一致。在GUMC-395的演化模型中,ecDNA由整合在MYC位点的串联化HPV基因组生成,随后经历了扩增和重组(Figure 5C)。这些HPV-宿主串联体通过次级重组和缺失事件继续演化(Figure 5C),最终形成了多样化但相关的变异结构,体现了异质链的特征(Figure 5B)。该模型为WGS数据中在多个片段连接处(包括F到G、H到I、J到K和K到L)观察到的CNV的逐步变化提供了可能的解释(Figure 5A)。研究者得出结论,HPV整合是GUMC-395中MYC基因高扩增的原因,这一关键事件可能促进了这种致命癌症的发展和生长。
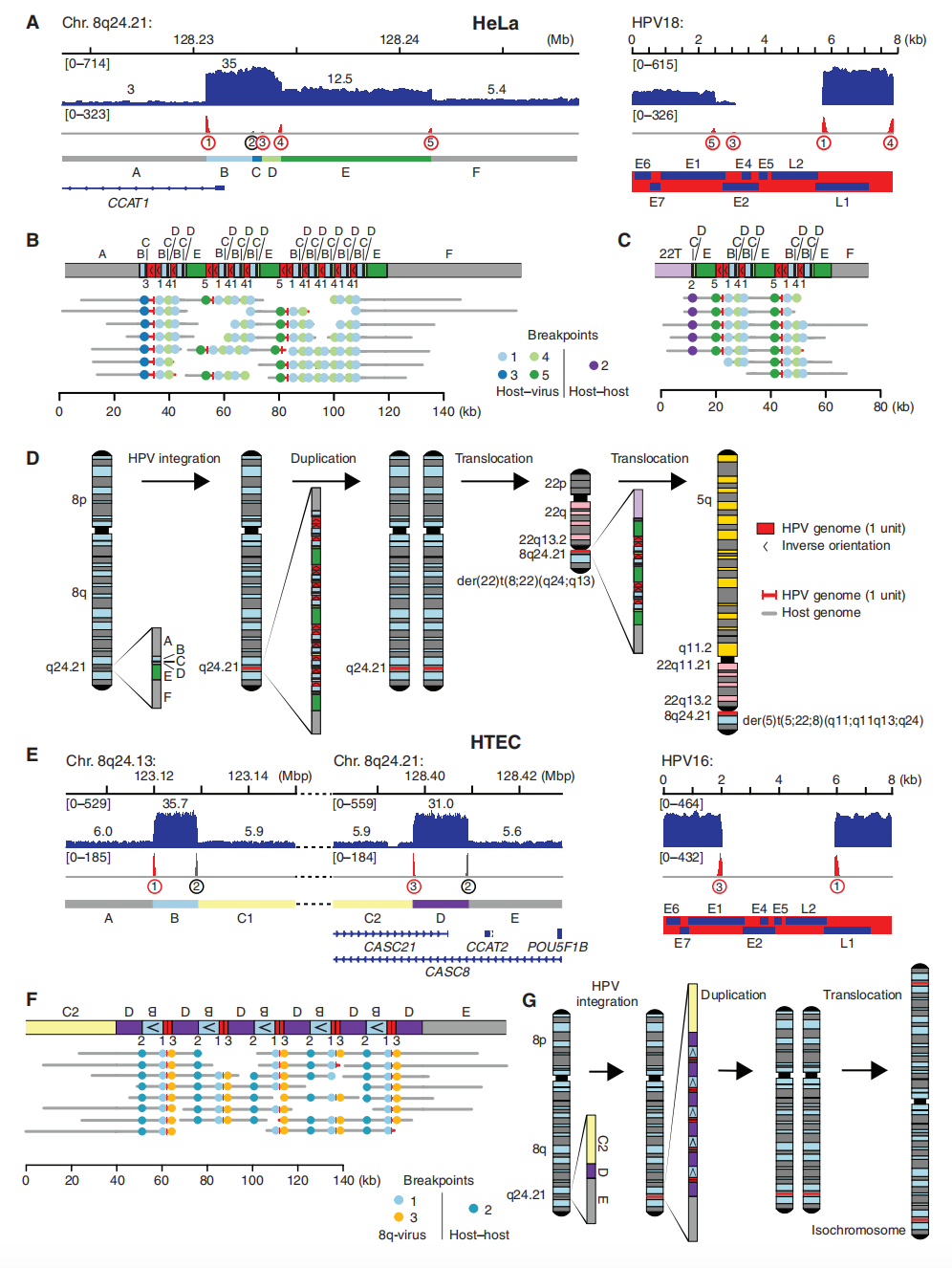
Figure 5. Intratumoral heterogeneity and clonal evolution are observed in LR-seq reads at MYC in GUMC-395 cells.
4.病毒-宿主串联体介导的染色体易位
通过荧光原位杂交(FISH)分析,使用HPV16探针检测GUMC-395细胞,发现病毒定位在所有中期分裂相中的两个8号染色体长臂(8q)拷贝和两个21号染色体拷贝上,这是由于涉及21号染色体着丝粒的t(8;21)(q24.21;q11.2)易位所致(Figure S5.1)。与此观察一致,LR-seq数据显示病毒-宿主串联体整合到8号染色体长臂24.21区(8q24.21)的宿主片段E附近(Figure 5B,group D1),以及第二个位点连接宿主片段E到8号染色体着丝粒(Figure 5B,group D9)。此外,检测到许多ONT读长连接了8号和21号染色体的着丝粒重复序列,跨越数千碱基。研究者推断这些串联体(可能以ecDNA形式存在)通过同源重组插入MYC位点,随后发生8号染色体复制、8号染色体长臂内倒位、t(8;21)(q24.21;q11.2)易位以及该易位的复制(Figure 5D)。
在HeLa细胞中,大量ONT读长支持病毒-宿主串联体整合到8号染色体长臂24.21区(8q24.21)MYC基因上游(Figure 6A-B),这与之前的分析结果一致。HeLa细胞中的t(8;22)(q24;q13)染色体易位最初通过光谱核型分析鉴定,但未通过WGS或单倍型解析数据检测到。其与HPV整合的关系(如果有)此前未报道。研究者的LR-seq数据独特地确认并解析了这一易位。研究者鉴定了病毒-宿主串联体,其断点与整合在8号染色体的断点相同,但连接了HeLa基因组片段C的5′端与22号染色体上2 kb的端粒重复序列(即5′-TTAGGG),形成断点2(Figure 6C)。与ONT数据一致,HPV18 FISH探针杂交到8号染色体的两个拷贝、t(8;22)(q24;q13)易位以及复杂的der(5)t(5;22;8)(q11;q11q13;q24)重排(Figure S5.1B)。WGS数据显示,8号染色体长臂的五个拷贝中有四个从HPV整合位点延伸到端粒。因此,研究者推断病毒-宿主串联体首先整合到8号染色体,随后发生8号染色体复制、易位到22号染色体端粒,以及从22号染色体着丝粒易位到5号染色体着丝粒(Figure 6D)。
总体而言,研究者对HeLa和GUMC-395细胞的综合分析表明,整合的病毒-宿主串联体不稳定,可以诱导染色体易位和其他形式的基因组SV。
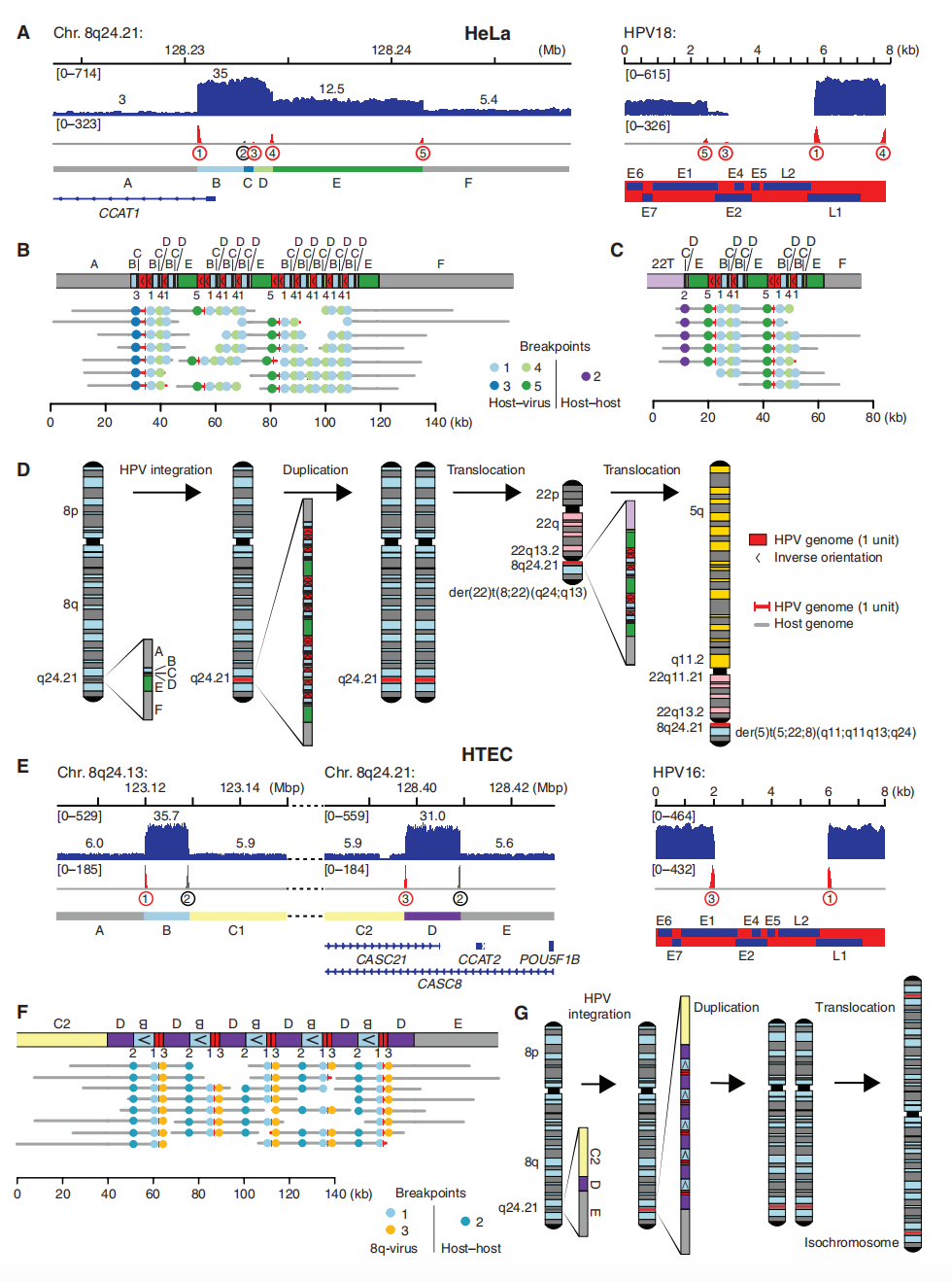
Figure 6. HPV integration in HeLa cells and HTECs induced CNV, SV, and intrachromosomal rearrangements.
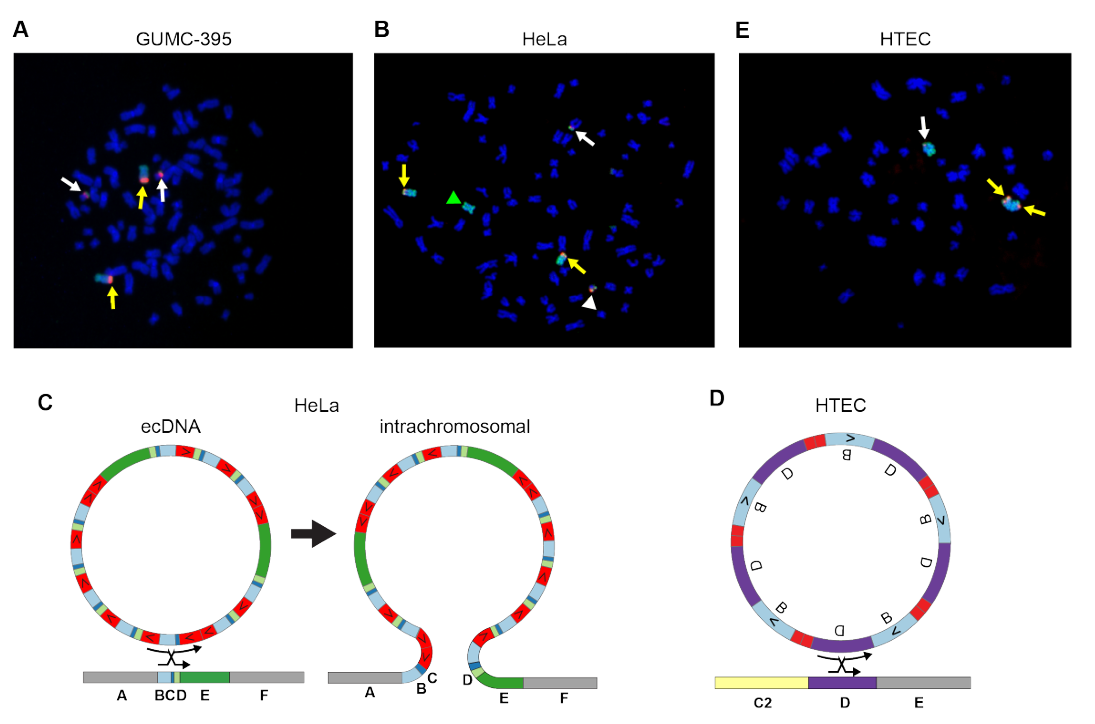
Figure S5.1. HPV integration into icDNA in GUMC-395, HeLa, and HTEC.
5.细胞系icDNA和ecDNA中的HPV整合
GUMC-395和HeLa的ONT数据支持病毒-宿主串联体整合到icDNA中。相比之下,VU147的ONT数据显示,包含17号染色体长臂12区(17q12)片段的病毒-宿主串联体以ecDNA形式存在,而病毒-宿主串联体则锚定在X染色体短臂21.1区(Xp21.1)的icDNA中(Figure S5.2)。为了评估GUMC-395、HeLa和VU147中病毒-宿主串联体以ecDNA形式存在的可能性,研究者使用HPV探针进行了中期荧光原位杂交(FISH)和Circle-seq分析。两种方法在所有检测的细胞系中均鉴定出含有HPV的ecDNA(Figure S5.3-S5.4)。GUMC-395和HeLa的Circle-seq数据与8号染色体长臂24.21区(8q24.21)MYC位点的扩增区域高度一致,支持ecDNA的存在,VU147中也观察到类似的数据。这一分析证实了细胞系中ecDNA和icDNA形式的结构相似的病毒-宿主串联体,表明其从染色体切除并插入染色体的过程。
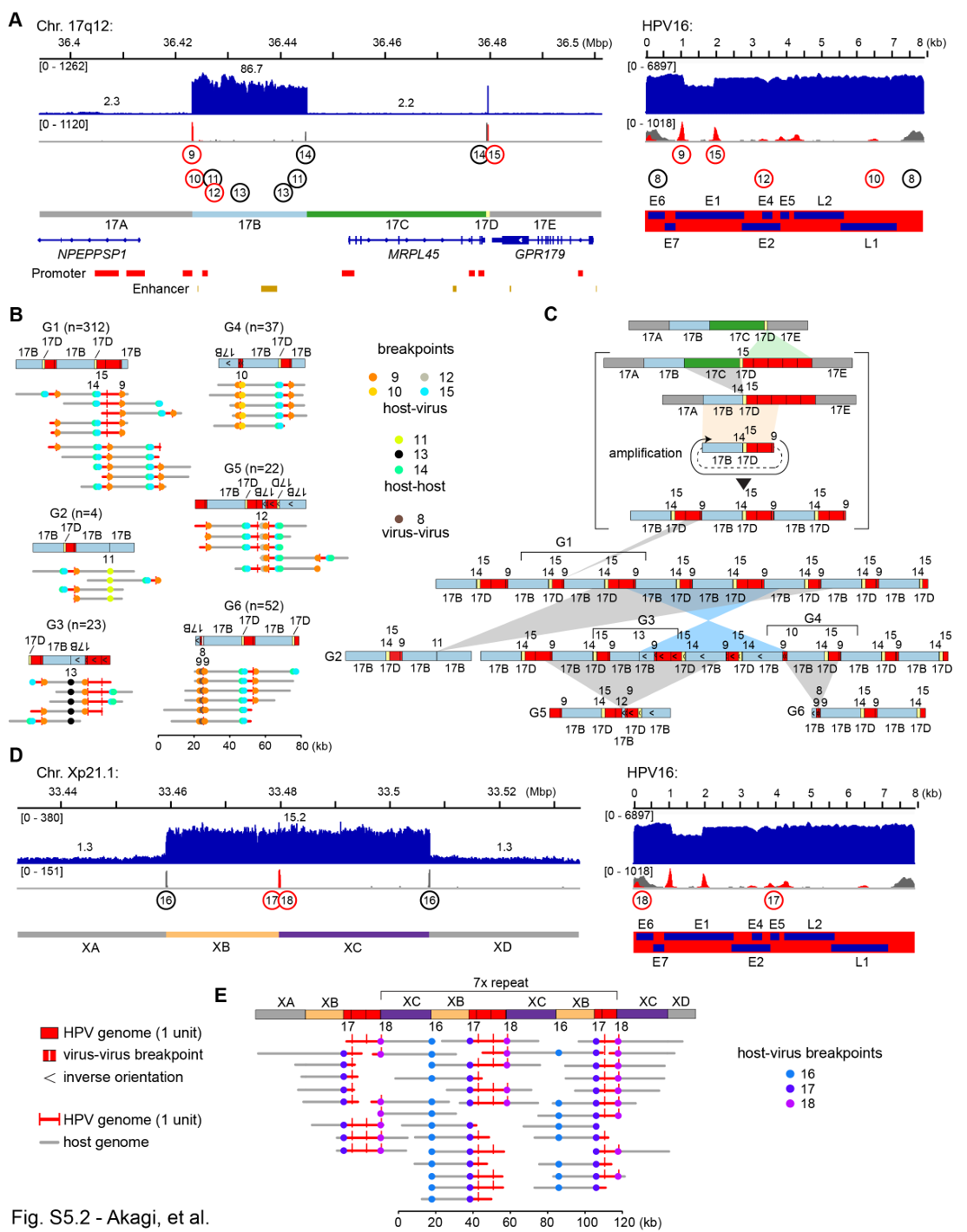
Figure S5.2. HPV integrants at Chr. 17 and Chr. X of VU147.
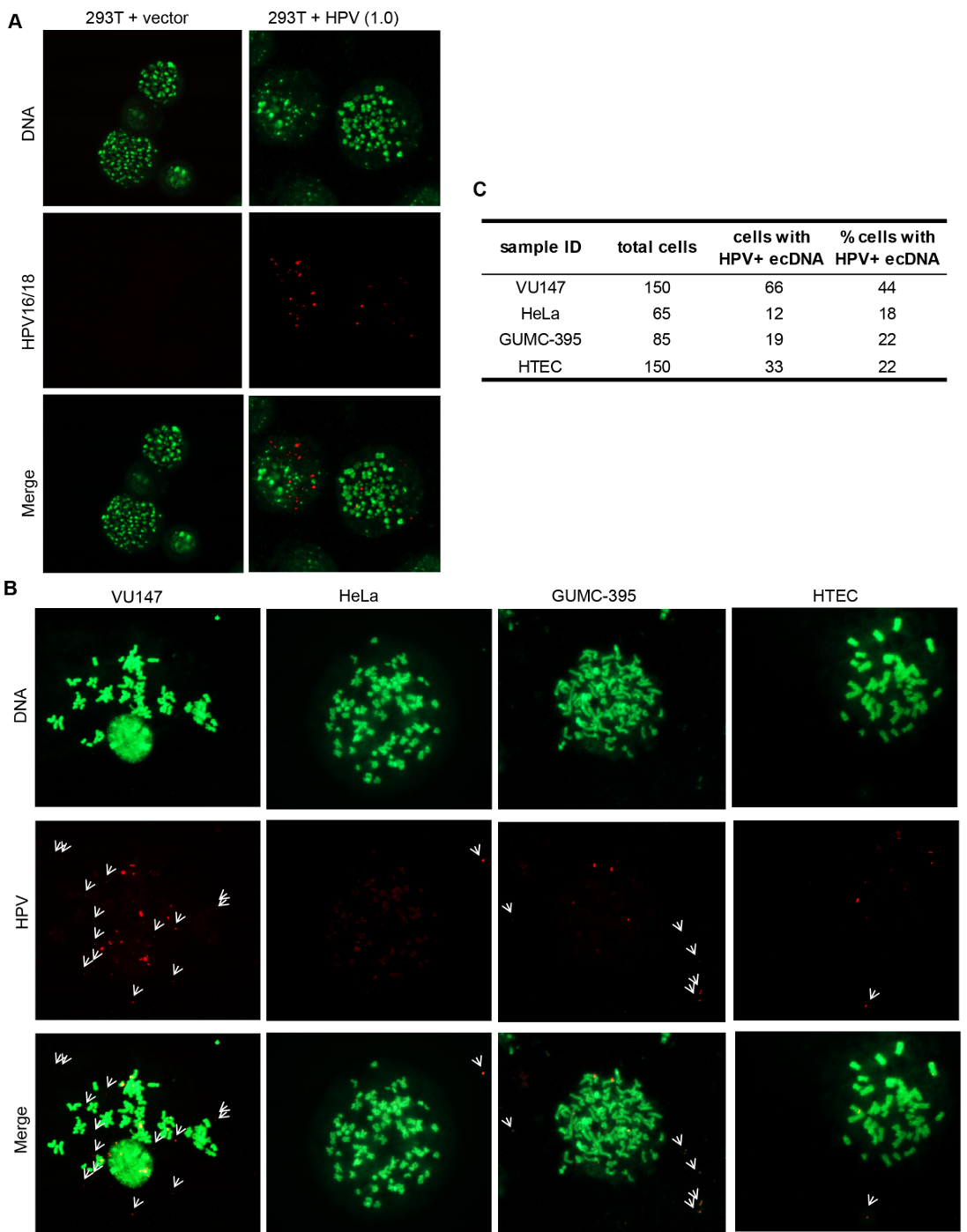
Figure S5.3. Metaphase FISH identifies HPV-containing ecDNAs in cell lines.
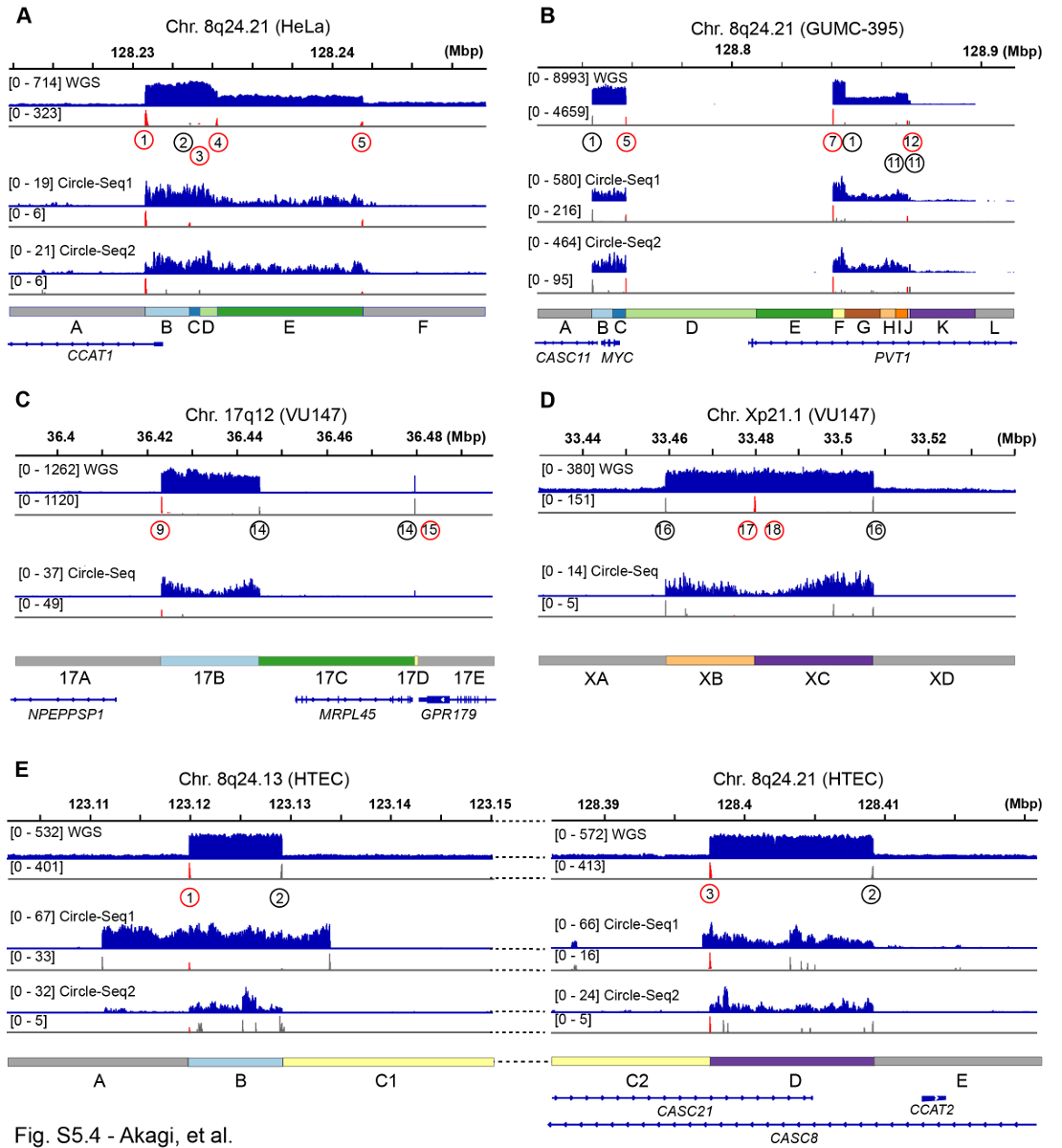
Figure S5.4. Circle-seq analysis of HeLa, GUMC-395, VU147, and HTEC cells.
6.HPV在MYC基因座的体外整合
人扁桃体上皮细胞(HTEC)系是通过将HPV16游离体DNA转染原代细胞并在体外进行克隆选择而建立的。病毒整合和HPV-宿主串联体的形成仅发生在体外细胞培养过程中。LR-seq数据显示,HTEC中HPV整合位点和MYC位点的基因组重排与GUMC-395和HeLa细胞中的情况具有显著相似性。在HTEC中,两个病毒-宿主断点位于MYC上游约350 kb的两个扩增基因组位点(即16–19n)的5′端(Figure 6E),其位置与HeLa类似(Figure 6A),SV与GUMC-395类似(Figure 5A)。ONT读长显示整合的病毒-宿主串联体与串联体中捕获的宿主DNA片段具有同源性(Figure 6F),支持由同源重组诱导的插入机制,与HeLa中的机制相似(Figure 6B;Figure S5.1C-D)。在连续传代的HTEC细胞中,与该位点对齐的Circle-seq读长显示出SV和其他不一致的重排,表明染色体内插入的不稳定性导致从该位点偶尔切除ecDNA(Figure S5.4E)。HPV16 FISH探针在所有检测的中期分裂相中定位到8号染色体长臂(8q)和等臂染色体i(8q)的两端(Figure S5.1E),表明在这些上皮细胞体外演化过程中,病毒整合先于这种染色体异常的形成(Figure 6G)。
7.异质链背景下的HPV基因组结构和转录本
几乎所有包含HPV序列的原发肿瘤和细胞系ONT读长都至少包含一个病毒复制起点(HPV16核苷酸7,838–7,906和1–100)以及编码E6和E7的区域(核苷酸83–858),即使其他HPV基因组序列被删除(或未观察到)。RNA-seq分析显示,在所有病例中E6和E7转录本水平均较高(Figure S6)。除了肿瘤5中E1被删除外,以ecDNA形式为主的病毒-宿主串联体的原发肿瘤含有全长HPV基因组,并表达E1和E2转录本。相比之下,以icDNA形式为主的病毒-宿主串联体的细胞系(即HeLa、GUMC-395和HTEC)在E1和/或E2中存在缺失,相应的转录本表达水平较低。因此,无论E2是否被破坏,E6和E7均持续表达。
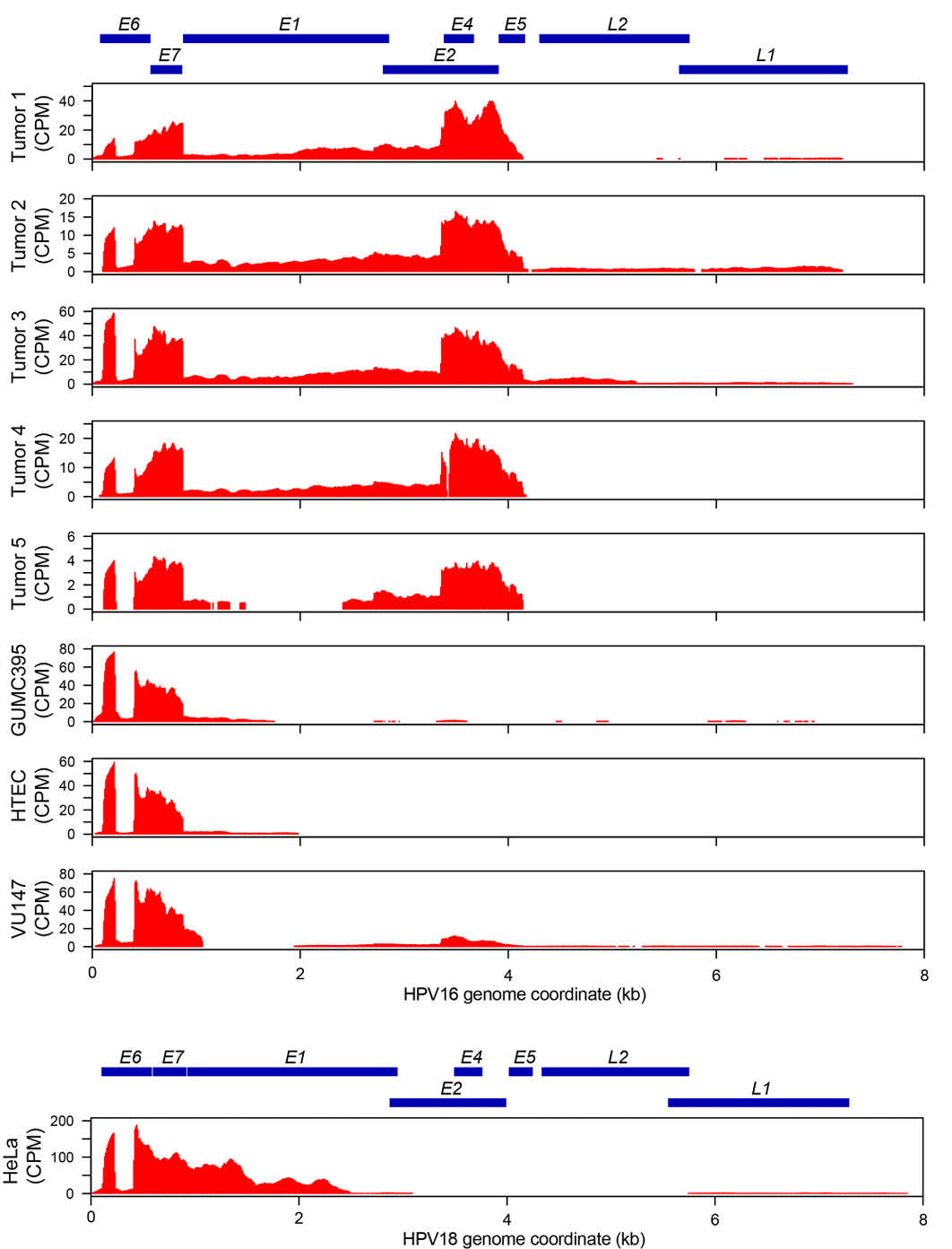
Figure S6. Transcription levels of HPV genes.
讨论
在本文中,研究者鉴定了一种由HPV整合在人类癌症中诱导的显著基因组SV形式——异质链,其特征是病毒和宿主DNA片段及断点的高度多样化、相互关联和重复模式,这些模式在肿瘤内共存。研究者在所有评估的癌症和细胞系中检测到了HPV相关ecDNA、icDNA或两者中异质链的强有力证据。基于LR-seq数据的进化模型将异质链解释为HPV整合诱导的异常宿主DNA复制和重组的结果,通常涉及串联化和环化的DNA。研究者推断,无论是ecDNA还是icDNA中的病毒-病毒和病毒-宿主基因组结构重排都具有不稳定性,从而导致进一步的肿瘤内异质性和克隆进化。因此,研究者也使用“异质链”这一术语来描述HPV整合诱导这种基因组异质性形式的逐步过程。
研究者此前对细胞系和原发肿瘤的WGS分析促使提出了一种“环化”机制模型,用以解释HPV整合位点观察到的广泛基因组SV。这个模型提出,HPV DNA的双链断裂促进了宿主DNA的捕获,导致插入断点,随后是病毒-宿主串联体的扩增、重组、修复和整合到icDNA中。然而,短读长WGS数据限制了研究者连接较长基因组距离的片段和断点的能力。通过新的发现,研究者扩展了这一HPV环化模型的内容,包括将不稳定的串联化HPV基因组生成并插入icDNA;在HPV ecDNA从icDNA切除并重新插入icDNA时捕获和重排宿主DNA;通过滚环复制或依赖重组的复制(RDR)扩增HPV-宿主片段;通过同源定向修复或复制过程中的模板切换在重复或同源片段之间发生重组,从而产生新的断点和片段模式;以及在串联体和端粒或着丝粒之间形成染色体倒位和易位(Figure 7)。
研究者进化模型中的步骤1至5(Figure 7)与现有文献一致。例如,Southern印迹和2D电泳提供了宫颈癌和细胞系中整合和/或游离体HPV串联体的低分辨率证据。通过对癌症基因组图谱(TCGA)的短读长WGS数据分析,提出了在未授权复制后从icDNA切除HPV整合体的可能性。在宫颈癌细胞系中,HPV E1-E2复合物与病毒复制起点的结合可以诱导未授权的DNA复制和基因组不稳定性。然而,研究者在这里观察到异质链在未检测到E1和E2表达的肿瘤和细胞系(如肿瘤5、GUMC和VU147)以及其他表达E1和E2的样本中均存在。尽管病毒-宿主串联体和混合游离体已被描述,但据研究者所知,异质链的发现及其特征(如Figure 7中步骤6至10所示)此前尚未被报道。
研究者注意到了异质链与其他导致癌症基因组SV的原因(如染色体碎裂、染色体编织、断裂-融合-桥循环(BFBC)和地震式扩增)之间的相似性和差异。异质链和染色体碎裂都与局部宿主CNV、SV和ecDNA的形成相关。尽管染色体碎裂以染色体片段随机重排为特征,但异质链中的病毒和宿主基因组片段以有组织、重复的模式连接。染色体碎裂ecDNA的形成涉及单一灾难性事件,而异质链则按顺序有序发生,通常涉及导致连续缺失和插入的重组事件。这种差异可能是由于HPV相关ecDNA在细胞分裂过程中被锚定在染色体上,而其他ecDNA则可能被排出微核。插入icDNA位点的病毒-宿主串联体共享由病毒基因组捕获的相同宿主DNA片段,表明同源重组介导了它们的整合。相比之下,染色体碎裂ecDNA优先整合在端粒附近。本文观察到的染色体易位在结构上比染色体编织更有序,后者涉及多个染色体的随机片段串联连接。异质链中的大规模倒位直接发生在端粒内,而BFBC事件则归因于端粒缺失。与地震式扩增类似,HPV串联体和异质链中的重排与CNV的逐步变化以及宿主基因(如MYC)表达增加相关。然而,地震式扩增中的CNV变化主要归因于重组,而研究者的模型表明异质链中连续缺失事件占主导地位,重组也可能起到一定作用。
癌症进化涉及两个基本过程:遗传变异和克隆选择。例如,通过比较LR-seq读长(如Figure 2B、4B和5B中的断点图所示),研究者展示了与HPV整合体直接相关的广泛肿瘤内基因组SV。研究者的进化模型表明,这些HPV整合体是每个肿瘤中单个细胞和亚克隆中异质链的诱导者。总体而言,研究者的数据和模型描述了HPV整合体选择包含宿主癌基因或其调控元件的DNA片段的过程(例如肿瘤5和细胞系GUMC-395及HeLa中的MYC),以及所有HPV阳性癌症中表达的病毒癌基因。HTEC中MYC位点的SV与肿瘤5、HeLa和GUMC-395中的相似性提供了强有力的实验证据,表明异质链是人类肿瘤进化中的驱动事件。
总体而言,与锚定在icDNA中的病毒-病毒和病毒-宿主串联体相比,ecDNA形式的串联体表现出更广泛的异质链,表明环状ecDNA是异质链中的活性介质或底物。在原发肿瘤中,几个受HPV整合影响的染色体位点缺乏LR-seq支持的icDNA锚定,表明它们主要以ecDNA形式存在。相比之下,细胞系的FISH分析显示,每个检测的细胞中HPV均整合在染色体DNA中。然而,细胞系的FISH、LR-seq和Circle-seq数据一致表明,整合的病毒-病毒和病毒-宿主串联体偶尔也会发生切除,形成HPV ecDNA。原发癌症和培养细胞之间的差异可能归因于不同细胞环境中ecDNA数量的差异。例如,复制和维持HPV ecDNA所需的关键因子可能在细胞系体外培养过程中下调或丢失。或者,携带icDNA HPV整合体的原发癌症亚克隆可能在细胞系衍生过程中获得选择性生长优势。
研究者注意到HPV相关ecDNA与神经母细胞瘤、胶质瘤和其他癌症中观察到的HPV阴性ecDNA之间的相似性和差异。后者ecDNA由非常大的(>1 Mbp)环状DNA组成,其复制机制尚不清楚。与HPV相关ecDNA类似,它们通常包含细胞癌基因(如MYC、突变EGFR)。这种ecDNA可以增加肿瘤内异质性,并促进快速适应选择性环境压力,归因于ecDNA在有丝分裂过程中在子细胞中的不均等复制和分离。相比之下,HPV相关ecDNA具有病毒复制起点并编码病毒蛋白(包括癌蛋白E6和E7)的特征。这些特征可能通过促进复制、分离和有丝分裂过程中与染色体的锚定来维持其稳定性。HPV相关ecDNA的丢失可能会受到强烈的负选择作用,因为E6和E7的表达是恶性表型所必需的。
HPV的复制模式主要取决于感染细胞的分化状态。在基底上皮细胞中,维持复制发生在S期,通过从病毒复制起点启动的双向θ复制进行,依赖于HPV E1解旋酶和E2转录调节蛋白。相比之下,滚环复制和RDR发生在G2-M期,较少依赖病毒复制起点,并且是单向的。后两种复制模式依赖于E7或E1诱导的ATM介导的DNA修复途径的激活。本文观察到的病毒-病毒和病毒-宿主串联体在单位连接处缺乏SNV或小的indel(Figure S3.1A-C),可能是由E6/E7表达、G1-S检查点的解除、细胞周期在G2期的延长停滞以及滚环复制或RDR引起的。
本文分析的每个原发癌症和细胞系都提供了一个时间快照,为研究者的异质链模型提供了信息(Figure 7)。研究者承认缺乏纵向收集的癌症和数据来验证事件的顺序。迄今为止,研究者尚未证明HPV ecDNA介导的宿主癌基因扩增直接促进癌症形成或进展。此外,尽管LR-seq数据相对于WGS数据具有许多优势(包括更长的读长分布和连续序列),但仍无法确定本文检测到的异质性、重复的病毒-宿主串联体结构是否连接在同一个非常长(>100 kb)的分子内,是否共存于同一细胞中、是否分布在不同的亚克隆中。研究者对多个ONT LR-seq读长验证的严格要求可能低估了每个癌症中分子异质性的程度。尽管研究者在所有研究的样本中观察到了异质链的证据,但需要更大的样本量来估计具有异质链的HPV阳性癌症的比例。
Figure 7所示的模型提出了HPV整合诱导CNV和SV形成、广泛多样性和异质链的机制。研究者得出结论,这种SV是由HPV整合引起的,并不反映HPV对预先存在的SV和CNV位点的偏好。这些数据扩展了研究者对HPV整合后果的理解,包括其在人类癌症中促进肿瘤内异质性和克隆进化的作用。此外,这些发现可能对其他整合到宿主基因组DNA中的DNA肿瘤病毒(如默克尔细胞多瘤病毒和乙型肝炎病毒)引起的癌症具有更广泛的意义。据研究者所知,迄今为止尚未使用LR-seq研究这些癌症的基因组结构或这些病毒诱导异质链的潜力。研究者推测,人类染色体内源性复制起点的异常启动也可能诱导各种形式的基因组不稳定性,可能包括异质链。
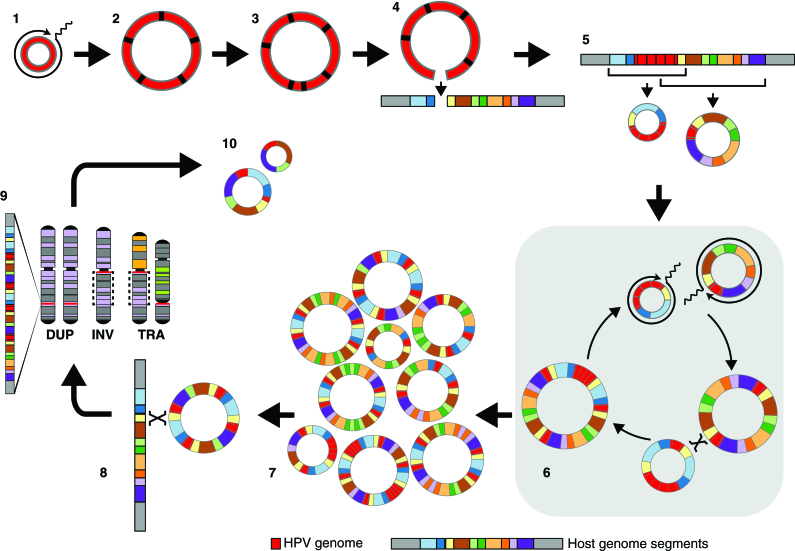
Figure 7. A model of HPV heterocateny development, depicting highly diverse but related genomic rearrangements including CNVs and SVs at HPV integration sites, is derived from multiple lines of evidence.

汇报人:胥飞宇
审核:张宇阳、陈欣、任建君
