


华西耳鼻喉学术前沿速递——文献精读(第55期)
精读分享│【Nature Communication】:空间转录组学揭示了预测生存和靶向治疗反应的独特和保守的肿瘤核心和边缘结构
英文题目:Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response
中文题目:空间转录组学揭示了预测生存和靶向治疗反应的独特和保守的肿瘤核心和边缘结构
期刊:Nature Communication(IF: 14.7)
单位:加拿大阿尔伯塔省卡尔加里市卡尔加里大学卡明医学院
发表时间:2023年08月
PMID: 37596273
PMCID: PMC10439131
DOI: 10.1038/s41467-023-40271-4

摘要
肿瘤微环境的空间结构对生物学特性和治疗反应有深远影响。在本研究中,作者对HPV阴性的口腔鳞状细胞癌(OSCC)进行了单细胞和空间转录组学的整合分析,以全面描述肿瘤核心(TC)和前沿(LE)区域的恶性细胞转录架构。结果显示,TC和LE区域具有独特的转录特征、相邻细胞组成以及配体-受体相互作用。作者发现,LE相关的基因表达特征在不同癌症中具有保守性,而TC则表现出组织特异性,这突显了肿瘤进展和侵袭的共通机制。
此外,作者发现LE基因特征与较差的临床结果相关,而TC基因特征与多种癌症类型中较好的预后相关。最后,利用计算模型,作者描述了OSCC中细胞发育的空间调控模式,这些模式与药物反应密切相关。作者的研究为跨癌种的TC和LE生物学提供了洞见,并提供了互动的空间图谱(http://www.pboselab.ca/spatial_OSCC/; http://www.pboselab.ca/dynamo_OSCC/),为开发新型靶向疗法奠定了基础。
关键词:口腔癌,癌症基因组学,肿瘤异质性
主要研究思路和方法:

介绍
口腔鳞状细胞癌(OSCC)是最常见的头颈部癌症,占据口腔黏膜上皮中发生的90%以上的癌症病例1–3。在印度等国家,OSCC是最常被诊断出的癌症4。多种致癌风险因素(如烟草和酒精使用以及HPV感染)与口腔癌的发生相关5。与其他头颈部癌症的亚部位(如口咽部)不同,HPV仅占OSCC病例的2–5%,目前HPV感染在OSCC中的意义尚不明确6。大部分OSCC病例为HPV阴性,主要由烟草和酒精使用驱动,改善此类病例的预后仍是一个尚未满足的需求5。尽管在过去几十年中对OSCC生物学的理解有所进展,但患者的生存率几乎没有明显改善;HPV阴性的OSCC患者中,生存超过5年的比例不足50%2。常规治疗方法如手术和细胞毒性化疗效果有限,且可能带来严重的并发症7,8,突显出基于生物学的替代治疗策略的必要性。
OSCC的侵袭和转移机制仍不清晰,但其占据了大部分与癌症相关的死亡9。超过50%的患者在治疗后的3年内出现局部复发或发生转移9。OSCC的前沿区域(LE),即位于OSCC肿瘤边缘的肿瘤细胞层,已被确认在临床分级中具有预后价值,并可能介导肿瘤的侵袭和转移10。然而,侵袭前沿及其他关键的空间特定区域的活性机制尚未完全了解11。以往对LE的研究主要通过免疫组织化学和原位杂交技术,因分析通量较低,难以全面描述OSCC的肿瘤微环境10,12。
最近的单细胞RNA测序(scRNA-seq)技术进展,使得在头颈部鳞状细胞癌(HNSCC)中探索肿瘤内异质性成为可能。例如,部分表达上皮间质转化(p-EMT)程序的肿瘤细胞位于前沿区域(LE),并表现出增强的侵袭能力13。相反,不表达p-EMT但表达上皮分化程序标记的肿瘤细胞则集中在肿瘤核心区(TC)13。然而,scRNA-seq研究最终缺乏将转录状态动态与肿瘤拓扑结构相关联的空间信息14。空间转录组学(ST)基于scRNA-seq技术,在提供基因表达数据的同时保留细胞的二维位置信息,全面展现了肿瘤微环境中转录异质性15。
在本研究中,作者利用空间转录组学(ST)和单细胞RNA测序(scRNA-seq)揭示了OSCC内部的转录异质性,通过确定和表征OSCC肿瘤结构的独特区域。作者发现位于肿瘤核心(TC)和前沿区域(LE)的恶性细胞具有独特的转录组特征和配体受体相互作用,这可能是由空间特异的癌细胞状态所致。作者还发现,TC和LE中保守的转录程序在OSCC及多种癌症类型中具有预后价值。通过预测性机器学习模型,作者观察到LE区域获得了多种癌症类型中共享的保守特征,而TC更具癌症类型特异性。此外,作者的研究利用RNA速率推断技术,识别了TC和LE中的肿瘤分化模式。最后,通过计算机模拟,作者识别出能够破坏OSCC患者TC至LE信息流的潜在有效药物。综合来看,作者的结果揭示了OSCC复杂的肿瘤景观,表明实体肿瘤可能采用一组保守且共同的机制进行进展和侵袭,这些机制可能成为治疗靶点。
结果
1、OSCC样本的空间转录组(ST)分析与细胞解卷积
作者使用10x Genomics Visium平台对来自10名患者的12个新鲜冷冻、手术切除的OSCC样本进行了ST分析(图1a和补充表1)。对24,876个位点的转录组进行了测序,每个位点在归一化后平均获得43,648条读取。数据经过归一化处理,校正了样本间的批次效应,并进行了降维以便于后续分析。每个肿瘤样本的苏木精-伊红(H&E)染色图像均由本研究的病理学家(M.H.)进行检查,并对形态学区域进行了标注(图1b和补充图1a–m)。
接下来,作者通过将ST数据与一个独立的、公开的HNSCC scRNA-seq数据集进行整合分析,确定病理学家标注的鳞状细胞癌区域中恶性肿瘤细胞和其他细胞亚群的组成。为了识别恶性肿瘤位点,作者严格将去卷积评分 >0.99(图1c)或CNV概率评分 >0.99(图1d)的细胞定义为恶性细胞。CNV分析显示在染色体3上存在反复缺失,而在染色体9上则有扩增(补充图1n)。根据设定的高可信度阈值,所有12个样本均被识别出含有空间去卷积或CNV推断的癌细胞,共鉴定出13950个恶性位点和10852个非恶性位点(图1e和补充图1o)。在对分类后的细胞进行批次效应校正和降维处理后,ST位点的统一流形近似和投影(UMAP)显示恶性位点呈散布分布,反映出转录谱的连续性(图1e)。使用标志基因LRRC15和GBJ2注释了HNSCC中保守的CAF亚型ecm-MYCAFs,使用ADH1B和GPX3注释了解毒型detox-iCAFs(图1f)16,17。空间去卷积分析显示,癌细胞、ecm-myCAF、中间成纤维细胞、解毒型iCAF、树突状细胞、肥大细胞、巨噬细胞和细胞毒性CD8+ T细胞在几乎所有样本中均有分布(图1f和补充图1p)。
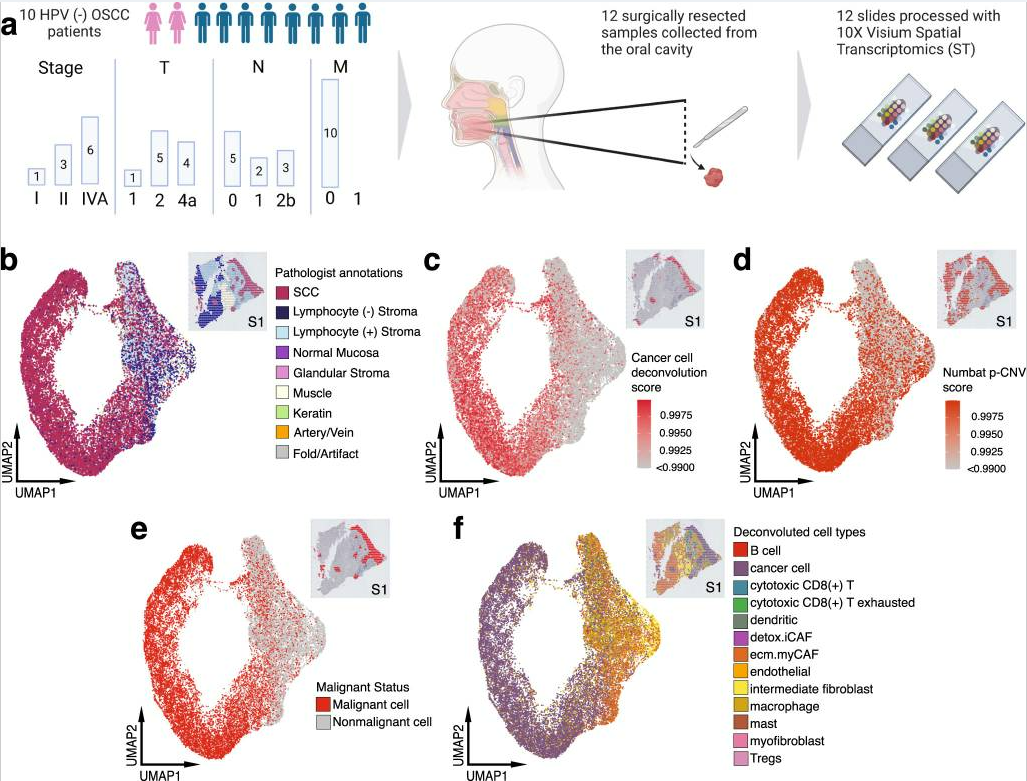
图1. OSCC患者样本的空间转录组(ST)分析和细胞解卷积实验设计概述。
a. 示意图展示了患者的临床数据以及样本获取和处理策略。包含来自所有12个空间分析样本的24,876个位点的UMAP投影,基于颜色编码。图由BioRender制作。b. 病理学家标注的区域,c. 基于Puram等人的scRNA-seq数据的单细胞HNSCC解卷积,d. 每个位点的CNV概率,e. 恶性位点状态,f. 基于解卷积和CNV概率的位点注释。HPV:人乳头瘤病毒,OSCC:口腔鳞状细胞癌,SCC:鳞状细胞癌,Tregs:调节性T细胞。
2、无监督聚类显示,肿瘤核心(TC)和前沿区域(LE)是肿瘤微环境中功能异质的成分
在识别并标注主要由癌细胞组成的恶性肿瘤位点后,作者进行了无监督的louvain聚类分析,以揭示癌细胞表达谱的空间异质性。在聚合的恶性位点中生成了14个louvain聚类,这些聚类可以划分为3个主要的聚类(图2a)。随后,作者通过差异基因表达分析(DGEA)对主要聚类进行了特征分析(图2b)。在聚类1中富集的主要差异基因(DEG)包括参与角质化的基因SPRR2D、SPRR2E、SPRR2A,以及抑制EMT的基因DEFB4A和LCN2(18,19);而聚类3的DEG则包括参与细胞外基质(ECM)结构的基因COL1A1、FN1、COL1A2、TIMP1、COL6A2(图2b和补充数据1)。聚类2的DEG则兼具聚类1和聚类3的特性,包括参与角质化的基因KRT6C、KRTDAP、KRT6B(20),以及ECM重塑的基因LYPD3、SLPI(21,22)(图2b和补充数据1)。有趣的是,CLDN4和SPRR1B作为HNSCC的TC标记基因13,及LAMC2和ITGA5作为HNSCC的LE标记基因13,分别对应于聚类1和聚类3(图2b、c和补充数据1)。这些发现促使作者将聚类1标注为“肿瘤核心”(TC),将聚类3标注为“前沿区域”(LE)。聚类2由于包含TC和LE的DEG特征而被标注为“过渡区”(图2d和补充图2a–m)。
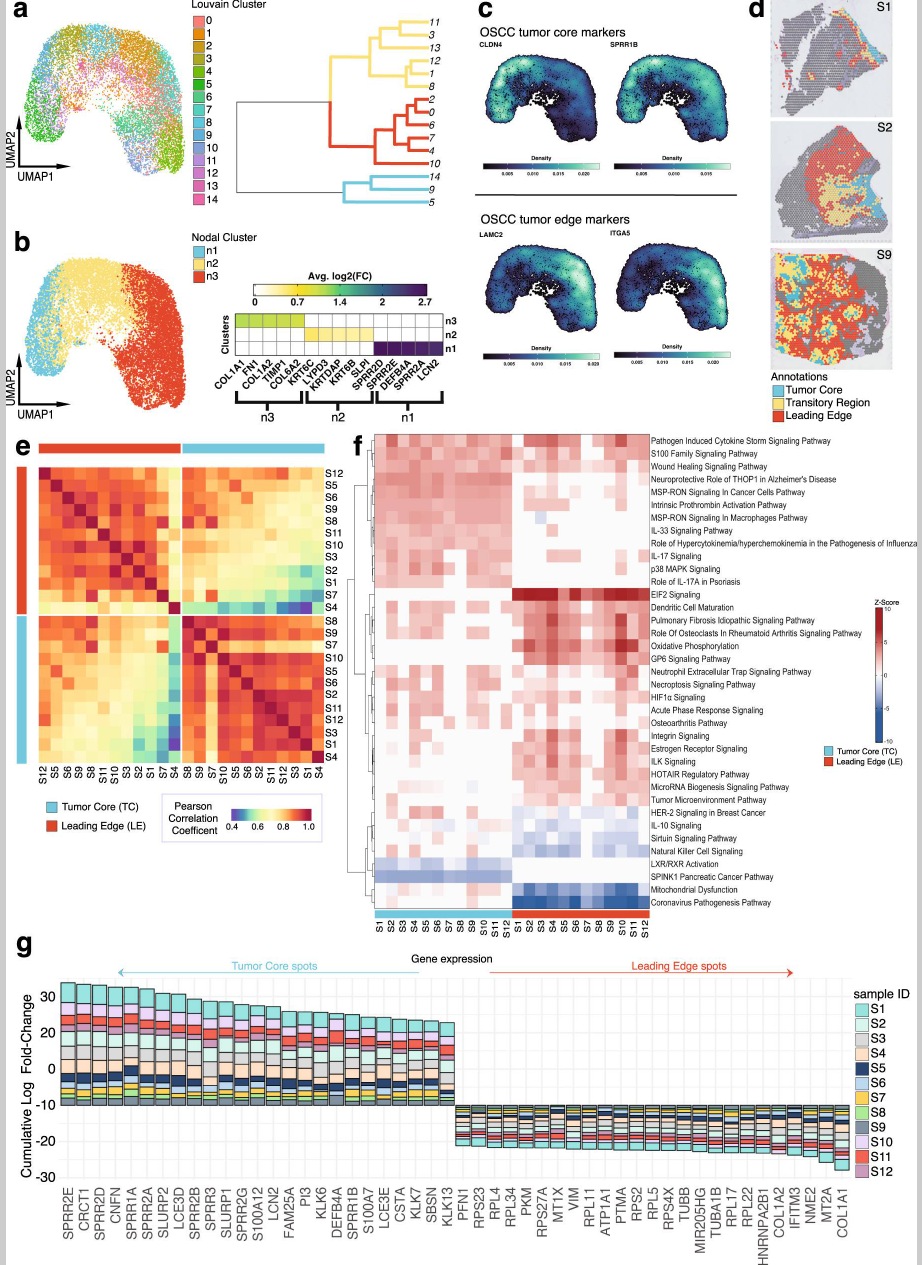
图2. TC和LE是OSCC微环境中的空间特异性区域。a. 来自所有12个空间分析样本的13,950个恶性位点的UMAP投影,基于Louvain聚类进行分区,附有展示聚类转录相似性的系统发育树。b. 来自所有12个空间分析样本的13,950个恶性位点的UMAP投影,基于三个主要节点聚类分区,附有显示每个聚类前5个差异基因(DEGs)log2(FC)的热图。c. Nebulosa核密度图显示了文献验证的OSCC肿瘤核心和前沿标记基因的表达情况。d. 样本1、2和9的TC、过渡区和LE注释。e. 所有空间分析样本中TC和LE注释的全转录组Pearson相关性热图。样本按转录组相似性排序。f. Ingenuity Pathway Analysis(IPA)热图,显示TC和LE通路的预测激活和抑制。显示的通路在10个或以上样本中被激活或抑制,并按每个样本中通路z评分的相似性排序。g. 共识图显示了在9个以上样本中显著差异表达的前25个基因的累积平均logFC(调整后的p值<0.001,双侧Wilcox秩和检验,Bonferroni校正)。相关来源数据已在“来源数据文件”中提供。
接着,作者探讨了LE和TC基因表达模式在不同患者间是否具有一致性。为此,生成了包含这两个空间区域的全转录组基因表达谱的相关矩阵(图2e)。观察到不同患者间的TC区域以及LE区域内部普遍具有高度的相关性。有趣的是,每个患者内部TC和LE表达模式之间的相关性相对较低,突显了这些肿瘤微环境区域的独特性。因此,作者的TC和LE标注代表了跨患者一致的独特转录组特征区域。
为探索TC和LE位点的功能差异,作者分析了这些区域内与癌症标志及关键癌基因通路相关的基因集的表达情况。结果显示,LE位点的细胞周期(p-adj < 0.001)、上皮-间质转化(EMT)(p-adj < 0.05)和血管生成相关基因表达较高(p-adj < 0.001)(补充图2n)。在LE中,EMT评分也表现出广泛的范围,与EMT连续性模型一致11。作者进一步分析了已发表的上皮分化和p-EMT基因集,发现这些程序分别定位于TC和LE位点(p-adj < 0.0001, p-adj < 0.001)(补充图2n)13。
TC中上调的细胞功能标志包括角质化、细胞分化以及抗菌和免疫相关通路,而在LE中上调了蛋白质翻译和核糖体相关通路(补充图2o和补充数据2)。Ingenuity Pathway Analysis(IPA)预测GP6、EIF2和HOTAIR的调控经典信号通路在LE中独特地激活(图2f)23–25。这些特征可能反映了OSCC LE在侵袭和转移行为中的作用。在TC中,作者观察到巨噬细胞中MSP-RON信号通路的激活、IL-33和p38 MAPK经典信号通路的激活,以及LXR/RXR和SPINK1经典信号通路的抑制(图2f)。这些发现表明OSCC的TC可能在肿瘤微环境中调节免疫反应,并在促进或抑制肿瘤进展中发挥作用26–30。
作者接着使用单细胞调控网络推断和聚类(SCENIC)分析,探索TC和LE之间的调控差异,以推断转录因子(TF)活性。SCENIC分析显示,在TC中上调了多个原癌基因TF EGR3和DLX5(31,32),以及抑癌TF MXI1、GRHL3和PITX1(33–35)(补充图2p和补充数据3)。相反,在LE中观察到多个TF的上调,包括调节细胞发育和分化的基因TP63和HOXB2(36,37),以及EMT调控基因CREB3L1、TCF4和NFATC4(38–40)(补充图2p和补充数据2)。
IPA上游调控分析也验证了在TC中原癌基因TF EHF和BCL3的激活(41,42),以及在LE中EMT调控基因SORL1和EGFR的激活(补充图2q)(43,44)。
TC和LE之间的差异表达(DE)分析在10个或以上样本中揭示了TC中有117个基因上调,LE中有91个基因上调(图2g和补充数据3)。与之前的HNSCC scRNA-seq研究相比13,作者的TC和LE DEGs分别仅与其中40个上皮分化基因和7个p-EMT基因重叠。TC中主要差异表达的基因包括参与角质化的基因SPRR2E、CRCT1、SPRR2D、CNFN和SPRR1A,而LE中主要基因则包括胶原蛋白(COL1A1、COL1A2),以及参与EMT启动和调控的基因MT2A、NME2、IFITM3(45–48),这表明LE中存在纤维血管生态位(图2g和补充数据4)。
3、TC和LE中存在不同的癌细胞状态
先前已有研究通过基因表达模式和临床结果,将HNSCC分为四种分子亚型,并由癌症基因组图谱(TCGA)验证49,50。为确定在OSCC的TC和LE中观察到的独特表达特征是否可归因于HNSCC的分子亚型组成,作者将TCGA的亚型表达特征与作者的ST数据集整合。在TC和LE中的位点根据与亚型表达特征的对应关系进行评分。结果表明,在多个患者的肿瘤微环境中可能存在多种分子亚型,但没有一致的亚型组成模式(补充图3a)。总体而言,TC状态中最富集的是Basal亚型(p < 0.0001)(补充图3b),而LE中最少的是Atypical亚型(p < 0.01)(补充图3c)。因此,仅基于基因表达对HNSCC患者进行亚型分类可能会过于简化这些癌症的复杂生物学特性。
接下来,作者通过使用Numbat包推断CNV事件,考察了肿瘤亚克隆结构与OSCC TC和LE的关联,分析克隆谱系和进化历史。作者发现,在整个OSCC肿瘤中存在多种亚克隆谱系,并且在TC和LE区域中亚克隆群体的比例相似(补充图3d)。这些发现表明,TC和LE之间的转录组差异不能完全由肿瘤内的基因多样性来解释。
随后,作者探讨了癌症干细胞(CSCs)的贡献是否可以帮助解释TC和LE表达谱的差异。CSCs是具有类似干细胞的祖细胞和恶性特性的癌细胞群体51。鉴于LE中丰富的EMT相关、转移和侵袭表达程序,作者假设CSCs可能更偏向于分布在LE。然而,作者发现LE和TC之间经典OSCC CSC标记的表达没有显著差异(p > 0.05)(图3a)。此外,UMAP投影中CSC标记的表达分布均匀,表明CSC群体在整个OSCC肿瘤中均有分布(图3a;密度图),并与非干细胞样的恶性细胞共存。
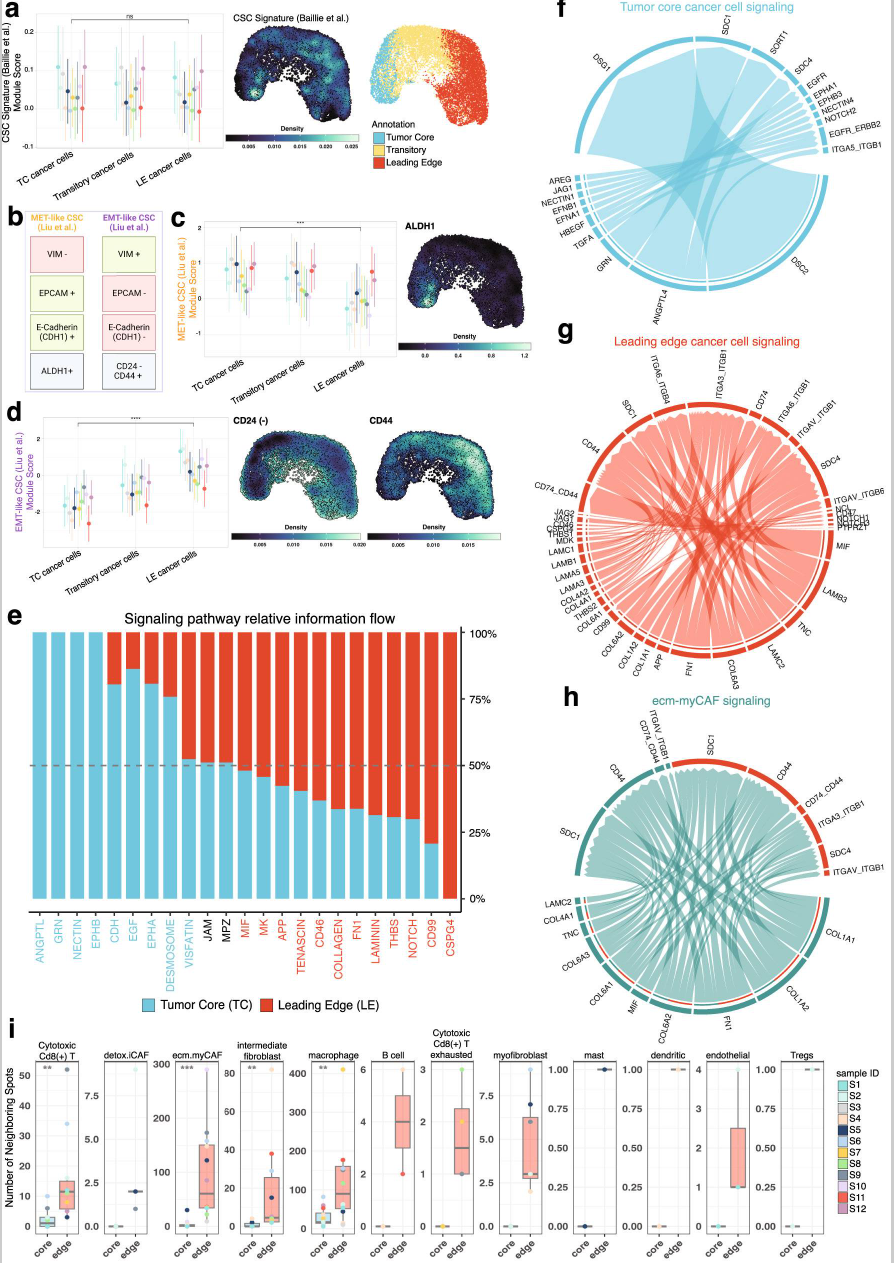
图3. TC和LE癌细胞状态是具有独特配体-受体相互作用的不同实体。a. 在TC、LE和其他鳞状细胞癌(SCC)位点中,CSC基因特征的表达比较,使用Nebulosa核密度图显示。提供TC、LE和过渡癌位点注释的UMAP以供空间参考。圆圈表示均值,线条表示标准差(n = 12个样本,来自10位独立患者)。b. Liu等人描述的上皮(eCSC)和间质癌症干细胞(mCSC)标记的示意图55。c, d. 使用Nebulosa核密度图比较TC、LE和其他SCC位点中mCSC(p值 = 2e−04)和eCSC(p值 = 1.5e−06)基因集的表达,显示mCSC和eCSC标记。圆圈表示均值,线条表示标准差(n = 12个生物学独立样本)。e. 堆叠条形图显示TC和LE位点中整体信息流富集的信号通路,路径以其在TC或LE中的主导性着色。f-h. 圆形图描述TC中的癌细胞、LE中的癌细胞以及LE中ecm-myCAF与癌细胞之间的细胞间相互作用配体-受体对,连接带的宽度表示配体-受体相互作用的强度。i. 比较直接邻接TC和LE位点的非癌性位点数量的箱线图。非癌性位点身份基于最富集的非癌性细胞类型解卷积确定。使用双侧Wilcoxon秩和检验和Benjamini–Hochberg FDR校正进行组间比较,*p < 0.05,**p < 0.01,***p < 0.001,***p < 0.0001。细胞毒性CD8 T细胞的p值 = 0.003,ecm.myCAF = 2.2e−04,中间成纤维细胞 = 0.002,巨噬细胞 = 0.008(n = 12个样本,来自10位独立患者)。箱线表示25%至75%分位数,中线表示中位数,须线延伸至1.5IQR范围内的最小值和最大值。相关数据在“来源数据文件”中提供。缩写:CSC 癌症干细胞,TC 肿瘤核心,LE 前沿区域,MET 间质-上皮转化,EMT 上皮-间质转化,CD24(−)逆向CD24基因表达(1/CD24),Tregs 调节性T细胞。
因此,作者认为TC和LE的独特生物学特征可由特异性癌细胞状态的存在来解释,这些状态包括保守的基因表达程序,这些程序在特定的肿瘤微环境相互作用下动态展现,涵盖了CSC和非干细胞样的恶性细胞53,54。先前的文献在探索CSC动态状态时提出,LE中可能存在类似间质的CSC,而TC中存在类似上皮的CSC(图3b)55。作者将这些不同的CSC状态相关的基因集应用于作者的ST数据集,结果证实LE中间质样CSC状态的表达较高(p < 0.001),而TC中上皮样CSC状态的表达较高(p < 0.001)(图3c, d)。通过连续组织切片的免疫荧光染色进一步验证了这些CSC状态的定位,结果显示CD24标记位于TC,而CD44标记位于LE(补充图3e)。这些发现进一步加强了TC和LE生态位内的可塑性,有助于转录上独特的癌细胞状态的传播。
4、TC和LE结构显示出不同的配体-受体相互作用
鉴于OSCC TC和LE的空间结构存在显著差异,作者进一步探讨了TC和LE中的细胞内和细胞外信号相互作用的性质和作用。作者使用CellChat包进行细胞间通信分析,从而得出细胞间通信网络的定量推论(补充数据5)。ANGPTL、GRN、NECTIN和EPHB信号通路仅在TC中观察到,而CSPG4信号通路仅在LE中观察到(图3e和补充图3f)。多种ECM重塑通路,包括胶原、Tenascin和Laminin,在TC和LE中均有表达(图3e和补充图3f)。与TC-TC信号相比,在LE-LE信号中上调的信号模式包括胶原、Laminin、Tenascin、FN1、MIF、APP、CD99、Notch和CSPG4通路(补充图3f),进一步证明了这些通路在促进癌症侵袭和转移中的作用。
接着,作者研究了恶性细胞之间的细胞间通信机制。由于ecm-myCAF在所有肿瘤样本中均有显著的细胞贡献(补充图3g),作者将其纳入分析。结果发现,ecm-myCAF表现出显著的细胞信号传导作用,与邻近的LE癌细胞相比,TC-TC和LE-LE之间的相互作用明显较少(补充图3g)。此外,ecm-myCAF与LE之间的相互作用强度远超LE-LE和TC-TC的癌细胞信号传导(补充图3g)。相较于LE-LE信号,ecm-myCAF-LE之间更多的相互作用数量和更强的作用力突显了ecm-myCAFs在LE处调控癌细胞行为的关键作用。
作者进一步探讨了代表TC-TC癌细胞、LE-LE癌细胞以及ecm-myCAF-LE癌细胞信号传导的特定配体-受体对。作者发现,TC-TC癌细胞可以通过粘附性配体-受体对DSC2-DSG1和ANGPTL4-SDC1等进行信号传导(图3f和补充数据5)。有趣的是,ANGPTL4-SDC1对可能抑制Wnt信号56,而Wnt信号是与癌症转移密切相关的主要通路之一57。
同样,LE-LE癌细胞可以通过粘附性配体-受体对LAMB3-ITGA6_ITGB4和LAMB3-ITGA6_ITGB1,以及炎性配体-受体对MIF-CD74_CD44进行信号传导(图3g和补充数据5)。MIF-CD74配体-受体对先前已被鉴定为启动癌基因信号通路58。
由于ecm-myCAF与LE癌细胞之间的高互动频率,作者还分析了这些信号通路。分析结果强烈预测了粘附性COL1A1-SDC1和FN1-SDC1配体-受体相互作用(图3h和补充数据5)。
为进一步表征肿瘤微环境,作者识别并量化了与恶性TC和LE位点相邻的非恶性位点数量。基于解卷积后富集的非癌细胞类型,作者将邻近的非恶性位点近似为特定的细胞类型。分析结果表明,相较于TC位点,LE位点邻近富集的细胞毒性CD8(+) T细胞(p-adj < 0.01)、ecm.myCAF(p-adj < 0.001)、中间成纤维细胞(p-adj < 0.01)和巨噬细胞(p-adj < 0.01)数量显著更高(图3i)。巨噬细胞是邻近TC的最丰富细胞类型(图3i),且与TC共同参与粘附斑和钙粘蛋白(CDH)信号通路(补充图3h)。巨噬细胞和细胞毒性CD8(+) T细胞还通过Laminin和胶原信号通路与LE和ecm.myCAF细胞类型参与细胞间信号传导(补充图3i),突显了它们在癌症信号中的重要作用。综上所述,作者的研究结果表明,TC和LE中的恶性细胞参与了不同模式的细胞间通信,进一步塑造了它们独特的生物特性。
5、TC和LE基因特征在跨癌症类型中具有保守性并具有不同的预后影响
由于作者标注的TC和LE区域在每个OSCC样本中均高度保守,作者进一步探讨了这些特异的分子程序是否也存在于其他癌症类型中。作者在TC位点、LE位点和其他剩余位点上训练了三个基于机器学习(ML)概率的预测模型,以生成空间区域预测模型(图4a)。接着,作者将该预测ML模型应用于30个不同类型的公开ST样本(涵盖17种癌症类型),以将每个样本中的各个位点分类为“TC”、“LE”、“过渡区”或“其他剩余位点”(图4b)。模型的10折交叉验证显示出稳健的表现(ROC值分别为:TC = 0.991,LE = 0.922,过渡区 = 0.943,其他剩余位点 = 0.958),其中LE区域的ROC值相对较低,这主要归因于其相对较低的敏感性(0.694)(图4c和补充表2)。
作者的分类器在空间上分离癌细胞状态表现良好,特别是在黑色素瘤(SKCM)、结直肠腺癌(COAD)、皮肤鳞状细胞癌(cSCC)样本以及宫颈鳞状细胞癌(CESC)样本中(图4d–h)。此外,作者的分类器在Abalo等人收集的四个连续的cSCC组织切片中产生了相对一致的注释结果(图4d和补充图4a),进一步强化了分类器结果的可重复性和可信度。作者在所有30个样本中识别出高度空间分离的LE位点(图4d–h和补充图4a)。儿童髓母细胞瘤和肝细胞癌切片显示出最低比例(1%)的推断前沿区域位点,这可能表明这些癌症类型与OSCC有显著差异(图4d)。同时,作者的ML算法在OSCC样本上训练后,在15/30个公开ST分析的切片中识别出空间分离的TC位点(图4d和补充图4a)。分类器在识别cSCC、黑色素瘤、CESC和COAD组织切片中的TC位点表现尤为出色,这可能归因于这些癌症中存在的角质化程序(图4d, g和补充图4a)。这些重要结果表明LE相关的表达状态在多种癌症背景中具有保守性,而TC相关的表达谱则更具组织特异性。
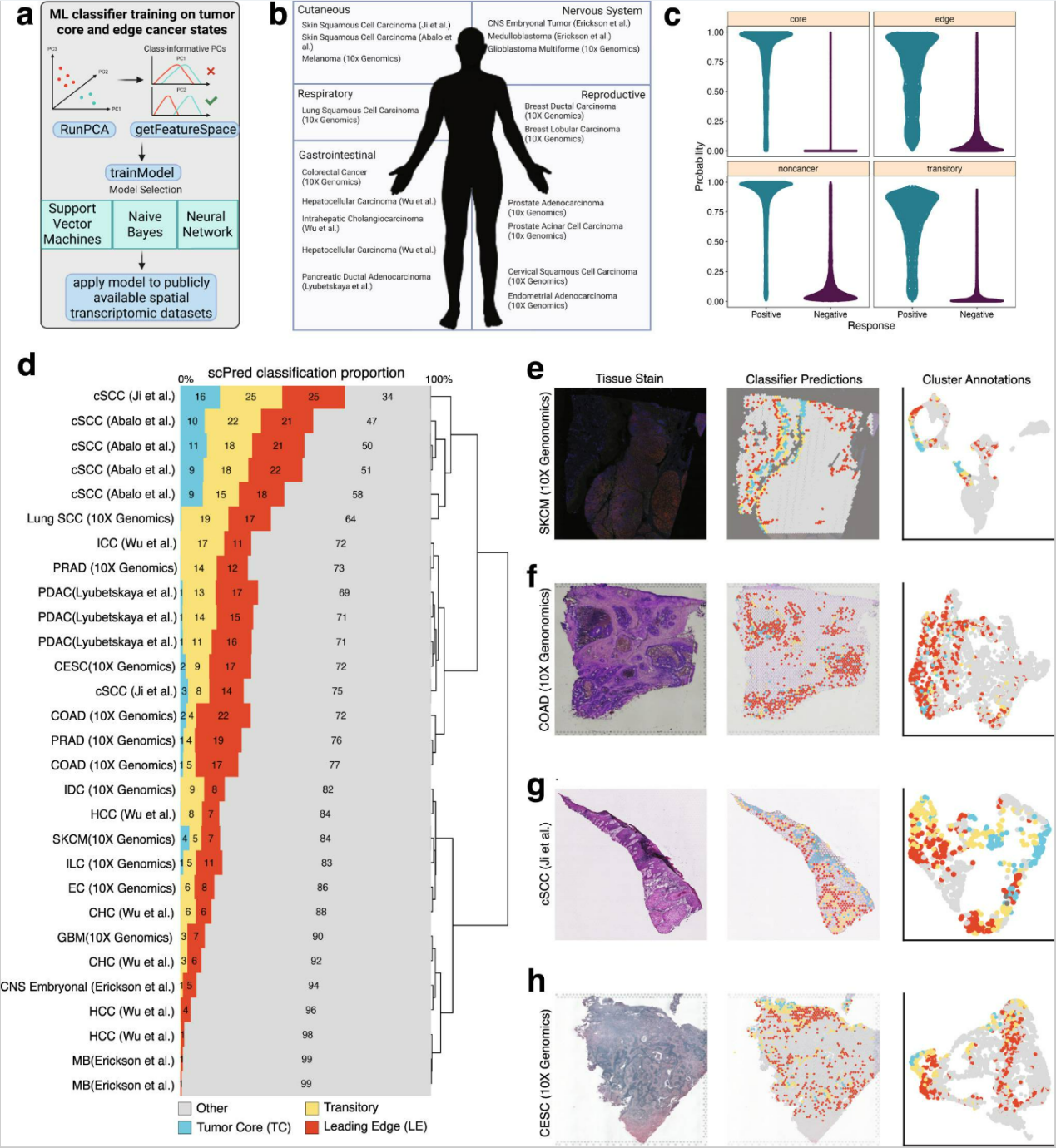
图4. 机器学习模型识别出多个癌症类型中保守的TC和LE特征。
a. 信息图描述了用于在公开的空间分析样本中识别TC和LE基因特征的机器学习策略。b. 信息图描述了包含在后续ML训练数据集中的公开空间分析样本59,101–104,110。图由BioRender制作。c. TC、LE、过渡区和其他区域的概率分布图。d. 条形图显示了每个空间区域在30个不同空间分析样本中的scPred分类评分。该图按TC、LE、过渡区和其他区域的预测比例相似性进行聚类。e–h. H&E染色组织切片(左),染色组织上的scPred投影(中),以及按scPred分类上色的UMAP(右),展示了cSCC、COAD和CESC代表性空间转录组测试数据集的结果。UMAP为统一流形近似投影。
缩写:cSCC 皮肤鳞状细胞癌,SCC 鳞状细胞癌,ICC 肝内胆管癌,PRAD 前列腺腺癌,PDAC 胰腺导管癌,CESC 宫颈鳞状细胞癌,COAD 结肠腺癌,IDC 浸润性导管癌,HCC 肝细胞癌,SKCM 皮肤黑色素瘤,ILC 浸润性小叶癌,EC 子宫内膜腺癌,CHC 肝细胞与胆管癌组合,GBM 多形性胶质母细胞瘤,CNS 胚胎中枢神经系统胚胎肿瘤,MB 髓母细胞瘤。
为探讨LE和TC特征的预后意义,作者将含有匹配生存数据的bulk转录组数据集(TCGA)样本纳入分析。共选择了275名HPV阴性OSCC患者进行生存分析,并根据TC和LE中差异表达基因的表达情况为每个样本分配了富集评分(见方法;图5a)。较高的TC单样本基因集评分与较低的病理分期显著相关(p < 0.05)(补充图5a),而LE评分与病理分期没有显著差异(p > 0.05)(补充图5b)。作者生成了Kaplan-Meier曲线,以可视化TC和LE特征高或低表达与总体生存(OS)、疾病特异性生存(DSS)和无进展生存期(PFI)的差异(图5b)。LE特征的高表达与OSCC患者较差的DSS(HR 0.60 (0.38–0.96, 95% CI]; p < 0.05)和PFI(HR 0.67 (0.45–0.98, 95% CI]; p < 0.05)相关,但与OS无显著相关性(HR 0.81 (0.56–1.16, 95% CI], p > 0.05)(图5b)。相反,TC特征的高表达与改善的OS(HR 1.51 (1.01–2.25, 95% CI]; p < 0.05)、DSS(HR 1.93 (1.09–3.41, 95% CI]; p < 0.05)和PFI(HR 1.82 (1.17–2.86, 95% CI]; p < 0.05)相关(图5b)。
为验证作者的结果,作者在一个独立的93名HPV阴性OSCC患者队列(GSE41613)中对TC和LE特征的高低表达进行了同样的比较。结果显示,TC特征在OS和DSS中表现出相似趋势,而LE特征的高表达与较差的OS和DSS相关(p < 0.05)(补充图5c)。TC和LE特征之间还显示出极弱的负相关(r = -0.17, p < 0.05)(补充图5d)。因此,作者的TC和LE特征在生存结局上表现出显著的相反趋势,可能是由于各自生态位中存在的不同机制所致。
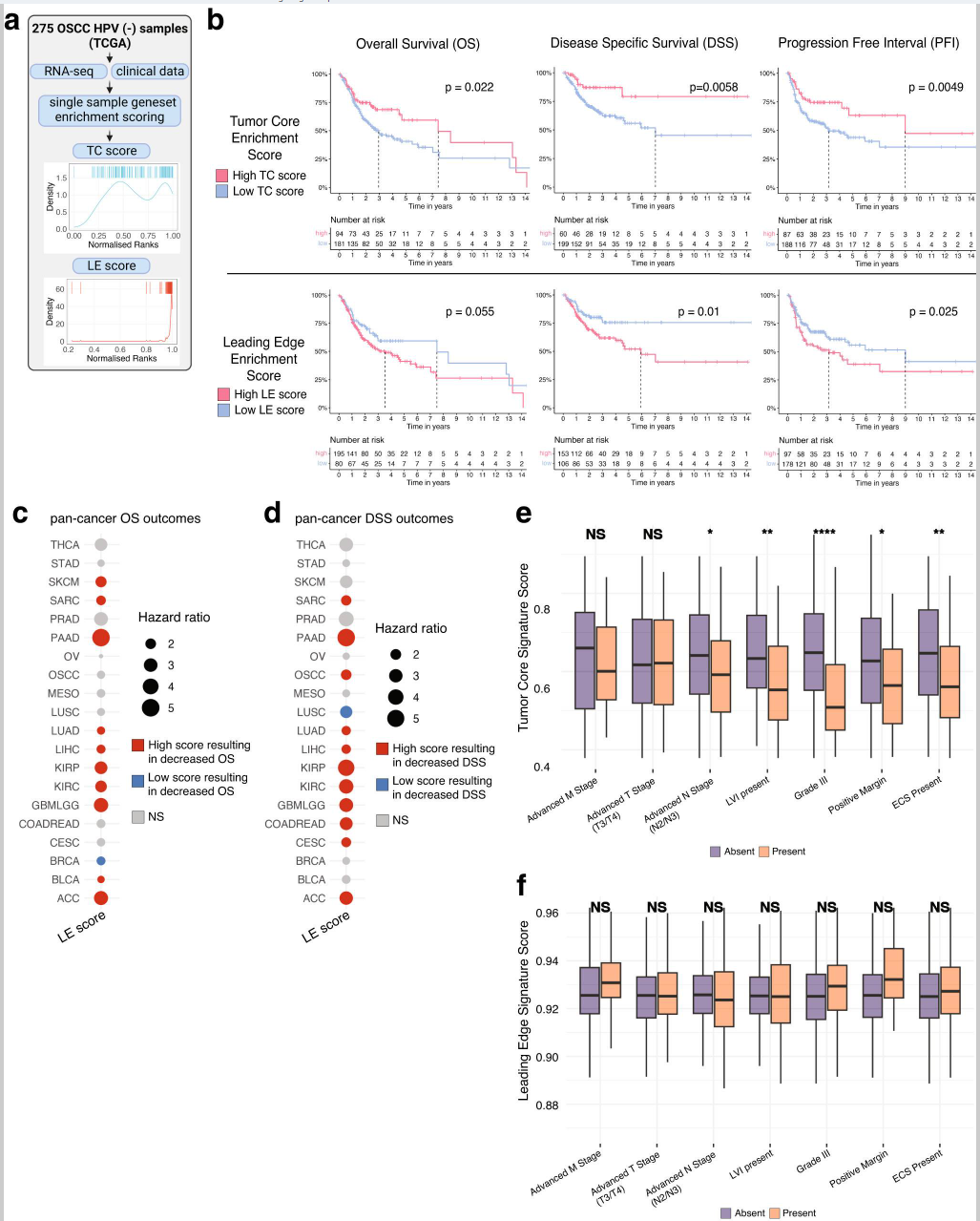
图5. TC和LE特征的生存关联和预后特性。
a. 信息图描述了在TCGA转录组数据中,TC和LE单样本基因集评分策略。b. 275例OSCC样本中根据TC(上图)和LE(下图)基因集富集评分分层的OS、DSS和PFI结局的Kaplan-Meier可视化图。显示的p值使用Cox比例风险回归计算。c, d. 从TCGA中20种常见癌症类型中获得的TC和LE基因集富集评分的OS和DSS跨癌症结局。p值和风险比使用Cox比例风险回归计算。e, f. 条形图显示与相关临床病理学协变量相比的相对TC和LE富集评分。显著性通过双侧Wilcoxon秩和检验并应用Benjamini–Hochberg FDR校正确定。误差条表示标准误(SEM)。*p < 0.05,**p < 0.01,***p < 0.001,***p < 0.0001。高级N分期p值 = 0.018,LVI = 0.002,III级p值 = 1.8e−06,阳性边缘p值 = 0.02,ECS = 0.002(n = 275个生物学独立HPV阴性OSCC样本)。箱线图表示25%至75%分位数,中线表示中位数,须线延伸至1.5IQR范围内的最小值和最大值。相关数据在“来源数据文件”中提供。
缩写:TC 肿瘤核心,LE 前沿区域,THCA 甲状腺癌,STAD 胃腺癌,SKCM 皮肤黑色素瘤,SARC 肉瘤,PRAD 前列腺腺癌,PAAD 胰腺导管癌,OV 卵巢浆液性囊腺癌,OSCC 口腔鳞状细胞癌,MESO 间皮瘤,LUSC 肺鳞状细胞癌,LUAD 肺腺癌,LIHC 肝细胞癌,KIRP 肾乳头状细胞癌,KIRC 肾透明细胞癌,GBMLGG 低级别胶质瘤和多形性胶质母细胞瘤,COADREAD 结肠腺癌/直肠腺癌,CESC 宫颈鳞状细胞癌,BRCA 乳腺浸润性癌,BLCA 膀胱尿路上皮癌,ACC 肾上腺皮质癌,LVI 淋巴血管侵犯,ECS 包膜外扩展。
由于LE状态在不同癌症类型中表现出一定的普遍性(图4d),作者进一步确定了LE特征的预后影响,并将分析扩展至TCGA中的20种常见实体肿瘤。结果表明,除乳腺癌(BRCA)的OS和肺鳞状细胞癌(LUSC)的DSS外,高LE评分在多种癌症中与较差的OS和DSS显著相关(图5c, d)。类似的关联模式也出现在LE与PFI之间,例外为黑色素瘤(SKCM)和LUSC(补充图5e)。
接着,作者分析了LE和TC特征与相关临床协变量之间的关联,以识别可能导致OSCC预后差异的因素。较低的TC特征评分与较高的淋巴结分期(p-adj < 0.05)、存在淋巴血管侵犯(p-adj < 0.01)、较高的肿瘤分级(p-adj < 0.001)、阳性边缘(p-adj < 0.05)和包膜外扩展(p-adj < 0.01)显著相关;而较高的LE特征评分则与任何临床特征均无显著关联(p-adj > 0.05)(图5e, f)。此外,还观察到EPIC CAF与LE特征富集评分之间存在弱负相关(r = -0.23, p < 0.05)(补充图5f),这表明除CAF活性外,可能还存在其他未探索的机制影响与LE相关的生存结局。
6、通过分析TC和LE之间的RNA剪接动力学揭示分化轨迹
RNA速度可以利用剪接和非剪接mRNA计数的比率预测单个细胞的短期未来状态60。将结果聚合在一起,有助于揭示癌细胞的发育轨迹,并识别负责转变的潜在驱动基因61。作者使用scVelo,它在RNA速度的基础上考虑了基因特异的转录动力学,来表征TC和LE中癌细胞的发育轨迹。在汇聚所有样本中空间解卷的癌细胞后,作者观察到一种从TC向LE延伸的分化层级(图6a)。该层级具有高度可重复性,且各位点之间高度一致,反映在所有位点上高于0.85的位点速度向量场置信度(图6a)。在单个患者层面上也观察到了类似的定向流模式(图6b)。
多个基因显示出推动TC向LE癌细胞状态分化的动态剪接行为(图6c)。TC和LE状态的主要候选驱动基因分别包括CSTA和IGHG3基因(图6c, d,补充图6a和补充数据6)。CSTA先前被鉴定为一个抑癌基因,涉及调控间质-上皮转化(MET)62。IGHG3是一个免疫球蛋白(Ig)基因,已在多种上皮癌中被检测到63,64。尽管确切机制尚不清楚,但肿瘤衍生的Igs被认为与肿瘤增殖、侵袭和转移、免疫逃逸以及介导EMT样表型相关联。这些发现预测TC经历了保留上皮样表型的增加分化,而LE则逐渐呈现出间质特性,这也与作者之前识别的CSC状态一致。TC和LE中其他差异剪接的驱动基因还包括原癌基因和抑癌基因(图6c,补充图6a和补充数据6)。
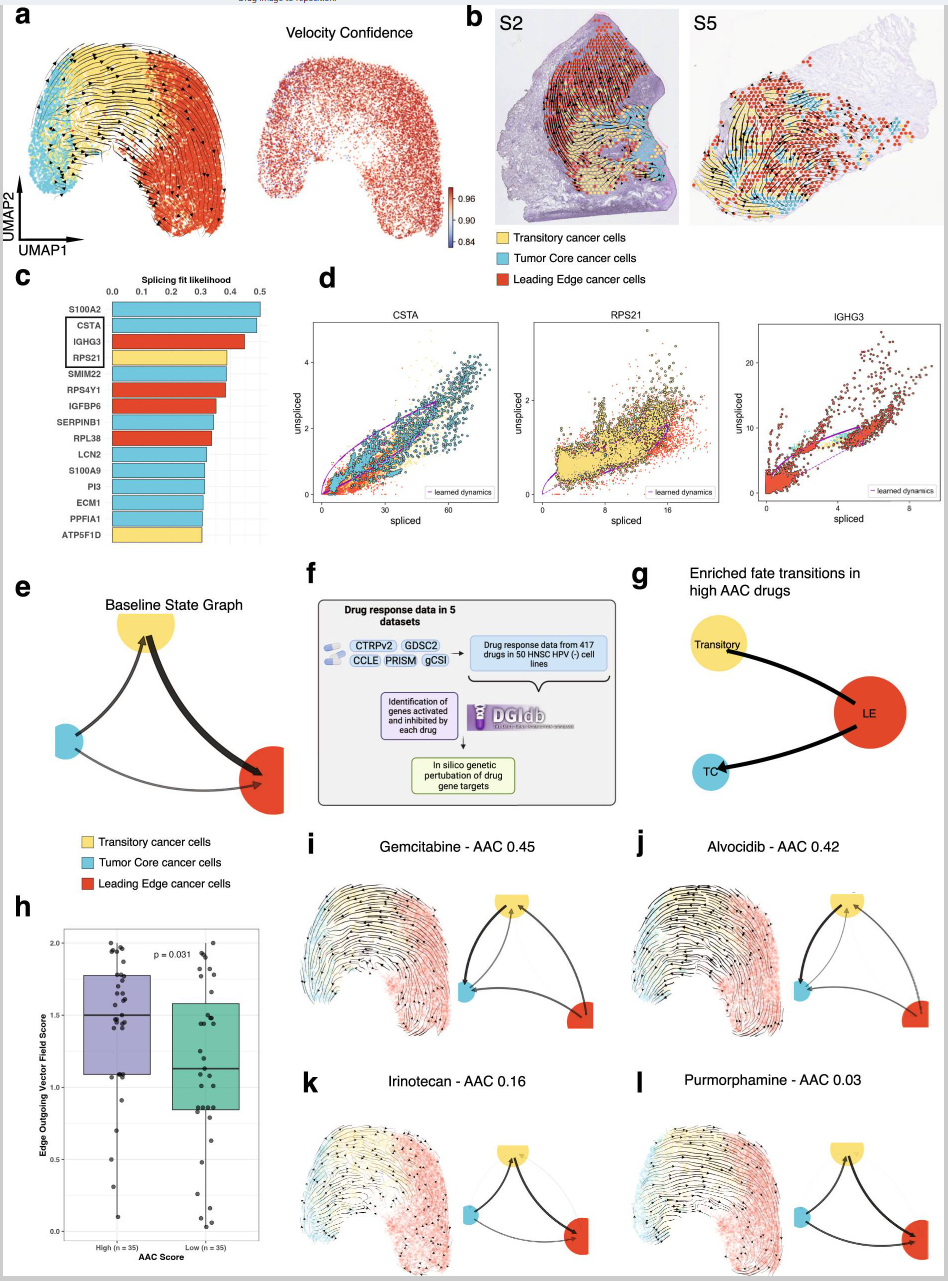
图6. RNA剪接动力学分析揭示了TC和LE中的差异化发育轨迹及治疗脆弱性。
a. 空间解卷的癌细胞位点的UMAP图,覆盖RNA速度流,以TC、过渡区和LE注释着色,并在UMAP图上叠加RNA速度置信度。b. 代表性空间分析样本(样本2和样本5),覆盖RNA速度流并以TC和LE癌细胞注释着色。c. 条形图显示TC和LE区域中差异剪接的主要基因(具有动态剪接行为的基因)。条形颜色表示基因在TC或LE中的剪接形式占比较高。d. 相位图展示差异剪接基因的剪接和非剪接RNA比率,紫色线条表示预测的剪接稳态。e. 基于向量场积分推断的TC、过渡区和LE注释的细胞命运转变概率状态图。f. 信息图描述系统收集药物响应数据并进行计算机模拟药物扰动的策略。图由BioRender制作。g. 信息图状态图显示在高AAC药物中富集的细胞命运转变。h. 箱线图比较高AAC和低AAC药物之间的向量场强度,基于中位数进行分层。AAC组使用双侧Wilcoxon秩和检验比较(n = 70种独立药物)。箱线表示25%至75%分位数,中线表示中位数,须线延伸至1.5*IQR范围内的最小值和最大值。i, j. UMAP展示在计算机模拟中扰动两个高效AAC抗癌药物靶标后的结果向量场和状态图。k, l. UMAP展示在计算机模拟中扰动两个低效AAC抗癌药物靶标后的结果向量场和状态图。
缩写:AAC 曲线下面积(area above curve)。
7、OSCC治疗靶点的开发
尽管诊断和治疗手段不断改进,OSCC仍表现出较高的疾病负担、死亡率和治疗相关的发病率65。现有的治疗方法,如手术、化疗和放疗,仅对少数患者有效,OSCC复发依然是主要的死亡原因65。此外,当前的治疗方法可能引起显著的发病率并降低生活质量(QoL),这与非特异性细胞死亡相关,包括局部缺损、言语和吞咽功能障碍及其他毒性副作用65,66。虽然靶向治疗在其他癌症中已能减轻毒性,但在OSCC中应用较少65,67。为解决OSCC中缺乏有效靶向疗法的问题,作者采用了计算机模拟扰动方法,以识别可能预测OSCC治疗成功的保守RNA速度模式(图6e, f)。Dynamo是一种计算机模拟技术,能够基于学习的剪接向量场准确预测基因扰动后的细胞命运转变68。
作者分析了PharmacoDB中的药物响应数据69,涉及至少25种HPV阴性HNSCC细胞系的417种药物,并发现140种药物具有DGIdb70识别的药物-基因相互作用(图6f和补充数据7)。接着,作者将分析限定在表现出显著扰动的70种药物上,并根据中位数(0.164;范围为0.026–0.560)将药物分为“高AAC”和“低AAC”组(补充数据7)。作者为肿瘤核心和前沿区域细胞间的“流入”和“流出”转变概率得出定量推论(图6e)。在有效药物(高AAC)中,基于Dynamo的计算机模拟扰动普遍显示出转变层级的基准状态逆转,即LE的流出转变概率增加(图6g)。这也反映在高AAC药物相较于低AAC药物显著增加的LE流出转变概率的定量测量中(p < 0.05)(图6h)。在个体药物层面上也复现了这些结果(图6i, j)。无效药物(低AAC)中则观察到相反的表型,表现出类似于基准状态的转变层级(图6k, l)。有趣的是,常见免疫治疗靶点(抗PD-1、抗CTLA-4)基于Dynamo的计算机模拟扰动显示出与有效药物(高AAC)类似的结果,以LE流出信号为主(补充图6b, c)。尽管有趋势,TC中的流入信号没有观察到显著差异(p > 0.05)(补充图6d);几个有效药物的异常值导致了显著性缺失。因此,有效药物可能通过诱导LE状态的逆转来表现其疗效,部分药物特异性地促进向TC状态的转变。药物类别分层分析进一步证实了这些结果,显示LE流出和TC流入转变概率的显著差异(补充图6e, f)。然而,某些药物类别的统计效能不足,难以得出确定的生物学推论(补充图6e, f和补充数据7)。
讨论
肿瘤内异质性是癌症患者治疗失败和生存率低下的主要决定因素之一71,72。然而,肿瘤微环境中异质性的空间基础仍然理解不足。尽管人们认识到TC和LE对肿瘤生物学具有深远影响,但这些区域的基因表达谱及其在形成异质性肿瘤微环境中的作用尚未得到系统探索。在本研究中,作者利用对12个OSCC组织样本的空间转录组学分析,全面表征了TC和LE的转录组特征,以深入揭示它们在OSCC的发生、发展和侵袭中的贡献。
首先,作者发现OSCC中的恶性TC和LE位点表现出不同的空间结构,具有跨患者保守的独特功能特征。TC具有角质化和分化状态,而LE则赋予了若干侵袭和转移特性。这些空间结构对于预后评估也具有重要价值——高LE特征评分和低TC评分与患者预后较差相关,而高TC评分具有保护作用,这可能部分归因于肿瘤分化的增加。有趣的是,这些空间结构在不同的癌症类型中具有一定的普适性。通过作者的机器学习模型,作者发现LE在多种癌症中具有保守性,且可以较高精度地进行注释,而TC中的特征可能仅限于具有相似起源的组织。此外,LE特征和TC特征在预后关联中表现出相似的趋势:LE特征在多种癌症中具有预后作用,而TC特征则相对特异,且预后作用仅限于少数癌症类型。总体而言,作者已识别出一个在多种癌症类型中具有保守性的LE相关转录程序,其与较差的患者预后相关。
作者还发现,TC和LE之间的基因表达差异并非由HNSCC的分子亚型组成或不同肿瘤进化克隆谱系的遗传差异所主导;作者的结果表明,在单个肿瘤样本中可能存在多种HNSCC亚型。作者提出,TC和LE的转录组差异由空间上独特的癌细胞状态驱动。通过RNA速度分析,作者发现这些空间独特的癌细胞状态由一个分化层级控制,从TC的类似祖细胞状态发展为LE的更具专一性的癌细胞状态。有趣的是,在分析CSC结构的细胞状态时,作者观察到了类似趋势:间质样CSC分布于LE,而上皮样CSC则位于TC。综上所述,作者认为TC状态的癌细胞可通过逐渐获得更具侵袭性的EMT样表型而转变为LE状态,进而促进癌症的侵袭和扩散。尽管已有多项研究描述了转录上独特的癌细胞状态13,53,73,74,作者的研究表明这些状态也受到空间调控。
最后,作者展示了将RNA速度应用于ST数据以预测药物响应的案例。作者发现有效药物(高AAC值)通常富集于引发LE状态逆转的流出转变概率,突显了LE作为一个重要治疗靶点的潜力。部分药物可能通过诱导癌细胞转变为TC状态来进一步提升其疗效。作者推测,有效的抗癌药物通过引导癌细胞从LE样状态转变为TC样状态,使这些细胞表现出较少的侵袭性,从而抑制侵袭/转移信号。Alvocidib是一种CDK抑制剂,目前正用于急性髓性淋巴瘤(AML)的研究75,在作者的分析中展示出LE流出和TC流入转变信号的高值。先前的研究显示CDK抑制剂在OSCC细胞系中具有良好效果76,77,Alvocidib或许是进一步研究的有前景候选药物78,79。重要的是,作者的扰动分析结果表明,空间转录组数据可用于计算机模拟评估药物疗效,并展示了LE作为可信药物靶点的潜力。
作者的工作旨在描述和揭示TC和LE之间独特的空间基因表达模式,代表了ST在OSCC中的应用。未来还存在许多激动人心的研究方向,包括深入理解癌症侵袭和转移的保守特征,以及指导以LE为靶点的泛癌治疗策略。作者相信,从本研究中获得的时空机制见解将有助于开发改善的OSCC靶向治疗方案,并为其他癌症的治疗提供参考。
方法
样本采集与标注
在阿尔伯塔癌症研究伦理委员会的批准下(研究ID:HREBA.CC-16-0644),从阿尔伯塔癌症研究生物样本库获得了新鲜冷冻的OSCC组织。所有患者在组织收集和研究中均提供了知情同意书,且未向参与者提供补偿。肿瘤样本在手术时收集,嵌入最佳切割温度(OCT)化合物中,并在−80°C冷冻保存,直至用于本研究。样本在冷冻切片机上切成10 µm厚的切片,进行苏木精和伊红(H&E)染色,并用于空间转录组(ST)分析。H&E切片用于由研究病理学家(M.H.)对组织进行标注。标注区域包括:鳞状细胞癌(SCC)、淋巴细胞阳性间质、淋巴细胞阴性间质、正常黏膜、腺体间质、肌肉、角质、动脉/静脉和人工痕迹。病理学标注从Loupe Browser(版本6)中导出,并导入Seurat进行进一步分析。
空间转录组分析
为了确定适用于OSCC组织的最佳透化时间,从而优化后续基因表达协议,对组织进行了优化处理。根据制造商的协议(10x Genomics,Pleasanton,加州,美国),使用Visium空间基因表达玻片和试剂盒(16次反应,目录编号:PN-1000184)在OSCC冷冻切片上进行了空间转录组学分析。简而言之,将嵌入OCT的10微米厚的OSCC冷冻切片置于Visium空间玻片上,进行24分钟的酶促透化。cDNA从与玻片上捕获引物结合的mRNA中生成,cDNA定量使用Agilent Bioanalyzer 2100(安捷伦技术公司,加州,美国)的Agilent Bioanalyzer高灵敏度试剂盒(目录编号:5067-4626)进行。cDNA文库在位于卡尔加里大学(阿尔伯塔省,加拿大)的健康基因组学与信息学中心(CHGI)通过Illumina NovaSeq 6000测序仪使用SP流动槽(200循环)进行测序。测序读取结果使用10x Genomics Space Ranger 1.3.1流水线比对到标准GRCh38参考基因组。12个样本通过比对QC后,使用10x Genomics Space Ranger的aggr功能聚合,以在样本间对读取深度进行标准化。聚合后的样本(n = 12)总共恢复了24,876个位点,每个位点的测序量在归一化后平均为43,648次读取。
空间转录组数据预处理
将聚合的HDF5矩阵导入R并按样本拆分。每个样本的特征条形码矩阵被导入R包“Seurat”(版本4.2.0)中进行标准化、质量控制、批次效应校正、降维和Louvain聚类80。表达少于200个特征的位点被排除在后续分析之外。样本级标准化使用“Seurat”(版本4.3.0)中的SCTransform函数进行。批次效应校正、整合和降维按照Seurat的单细胞RNA-seq整合指南(版本4.3.0)(https://satijalab.org/seurat/articles/integration_introduction.html)执行,无任何偏差。
细胞类型解卷积与恶性标注
从GSE103322下载单细胞HNSCC数据并使用“Seurat”(版本4.3.0)中的‘CreateSeuratObject’函数导入R。简要来说,单细胞数据使用‘SCTransform’进行标准化,降维处理并进行了Louvain聚类。根据原始文献中描述的标志基因13,将成纤维细胞亚型分配至Louvain聚类;ACTA2+TAGLN+细胞标注为肌成纤维细胞,CXCL1+PDPN1+ACTA2- MMP1-细胞则标注为中间成纤维细胞。在HNSCC中保守的CAF亚型使用标志基因LRRC15和GJB2标注为ecm-MYCAFs,ADH1B和GPX3标注为解毒型iCAFs16。‘CARD’ R包(版本1.0)81用于通过‘CARD_deconvolution’函数将空间转录组位点解卷为细胞类型。GSE103322单细胞数据集用作参考,空间转录组数据作为查询数据集。解卷的细胞类型数据被添加到Seurat对象元数据中以进行后续分析。
使用‘numbat’ R包(版本1.1.0)82对所有12个空间转录组对象进行分型感知的CNV推断,基于原始ST BAM文件。CNV推断按照numbat的空间转录组指南进行(https://kharchenkolab.github.io/numbat),无偏差。突变与正常概率(p_cnv)从numbat中导出并添加至‘Seurat’的元数据中以便后续分析。
如果位点的‘CARD’解卷比例超过0.99,或基于numbat CNV的突变与正常概率(p_cnv)超过0.99,则标注为“癌细胞”。此外,标注为“癌细胞”的位点还必须由研究病理学家标注为包含鳞状细胞癌。未标注为“癌细胞”的位点则基于CARD推断的最大非癌细胞解卷比例标注为对应细胞类型。
TC、过渡区和LE标注
为研究癌细胞异质性,作者仅对所有ST玻片中的癌细胞进行重新分析,使用“Seurat”(版本4.3.0)进行标准化、批次效应校正、降维和分辨率为1.0的Louvain聚类。为识别癌细胞身份的层级状态,在“Seurat”(版本4.3.0)中使用‘BuildClusterTree’函数构建系统发育树。此树通过ape R包(版本5.6-2)83中的‘plot.phylo’函数进行可视化,揭示出ST癌细胞的三个层级分区,随后在“Seurat”(版本4.3.0)中标注并可视化。使用‘FindAllMarkers’ Seurat函数(版本4.3.0)对三个节点聚类进行广泛的差异基因表达分析,该函数使用双侧Wilcoxon秩和检验并进行Bonferroni校正。差异表达基因的可视化通过‘SCpubr’ R包(版本1.1.2.9000)84进行。根据差异表达基因和前沿标记基因LAMC2和ITGA5,以及肿瘤核心标记基因CLDN4和SPRR1B的局部表达,将节点聚类标注为‘edge’(前沿)、‘core’(核心)和‘transitory’(过渡区),这些标记基因先前已在头颈癌中得到验证13。标记基因的核密度图通过‘SCpubr’ R包(版本1.1.2.9000)84中的‘do_NebulosaPlot’函数绘制,并使用R包‘Nebulosa’(版本1.8.0)85。
差异表达分析、共识绘图与相关性热图
使用Seurat(版本4.3.0)中的双侧Wilcoxon秩和检验并进行Bonferroni校正识别差异表达基因,设定logFC > 0.25和调整后的p值 < 0.001。利用构建共识函数(constructConsensus)86的改编版生成累积表达log2FC的堆叠条形图,展示每个基因在所有样本中的表达情况。共识图用于显示在超过9个样本(>9/12)中差异表达的前25个基因。每个样本的差异表达基因被导入Ingenuity Pathway Analysis(IPA)进行通路富集分析。IPA的导出结果被导入multienrichjam R包(版本0.0.57.9)(https://github.com/jmw86069/multienrichjam)。若通路在10个或以上样本中被激活或抑制,则使用multienrichjam的mem_enrichment_heatmap函数创建跨样本的通路富集图。整体转录组平均表达通过R包‘stats’(版本4.2.2)中的‘cor’函数进行Pearson相关性分析。相关性值通过‘ComplexHeatmap’ R包(版本2.14.0)87中的Heatmap函数进行可视化。
样本组间评分比较的统计方法
通过修改‘SCPA’ R包(版本1.2.0)88的代码识别TC和LE癌细胞状态间差异调控的标志通路。‘compare_seurat’函数查询包含集成数据的‘Seurat’(版本4.3)R对象中的标志基因集,并使用多变量分布测试检验差异性通路活性,同时应用Bonferroni校正。用于比较标志基因集的图表通过‘ggplot2’(版本3.4.0)和‘ggrepl’ R包(版本0.9.2)生成。
在每个位点中,使用Seurat(版本4.3.0)的‘addmodulescore’函数为特定的癌症标志基因特征进行评分。为检验TC和LE之间模块评分的差异,使用‘ggpubr’(版本0.5.0)函数‘stat_compare_means’进行双侧配对Wilcoxon秩和检验,并进行了Bonferroni校正。为广泛的CSC、上皮CSC和间质CSC状态特征的评分,使用Seurat(版本4.3.0)函数‘addmodulescore’构建表达模块。CSC基因特征根据文献中其预期上调或下调进行识别55。预测上调和下调的基因评分贡献随后累加以得出模块评分。模块评分的核密度图使用‘SCpubr’ R包(版本1.1.2.9000)中的‘do_NebulosaPlot’函数绘制,并使用R包‘Nebulosa’(版本1.8.0)进行可视化。
基因调控网络推断
使用SCENIC89管道的Python实现(pySCENIC版本0.12.1)90推断转录因子与其靶基因之间的调控相互作用。核心和前沿癌细胞数据按照pySCENIC教程(https://pyscenic.readthedocs.io/en/latest/index.html)中的默认和推荐参数进行处理,并使用hg19 RcisTarget数据库对调控模块进行修剪。pySCENIC输出的调控模块活性评分通过‘CreateAssayObject’函数添加到‘Seurat’(版本4.3)对象中。累积每个转录因子在所有样本中的AUCell评分logFC的堆叠条形图,通过改编构建共识函数并设定调整后的p值 < 0.05生成。
免疫荧光和基于荧光的细胞分析
将OCT冷冻组织切成6μm厚的切片并置于玻片上。切片在室温下风干30分钟,随后使用4%多聚甲醛固定15分钟。切片用1X PBS简短清洗后,用10%马血清阻断1小时。配制抗体稀释缓冲液,包括PBS、1% BSA、0.1%冷鱼皮明胶和0.1% Triton X-100,并对切片进行间接免疫荧光染色,使用兔多克隆抗CD24抗体(Abcam:1:250稀释;目录编号:ab244478)和鼠单克隆抗CD44抗体(IM7,Invitrogen:1:250稀释;目录编号:14-0441-82)作为一抗。敲除验证由制造商提供91,92,稀释规格按制造商协议选择。之后,加入相应的荧光团标记的二抗:Alexa 546标记的山羊抗兔抗体和Alexa 488标记的驴抗鼠抗体(1:500;Jackson ImmunoResearch Laboratories),以及DNA荧光染料Hoechst 33342(1:1000)以可视化细胞核。
为盖上盖玻片,使用Fluoromount-G(SouthernBiotech;目录编号:0100–01),并使用荧光显微镜在10倍放大倍率下捕捉组织切片的荧光图像(Zeiss AxioObserver Z1)。所有样本中CD24(2秒)、CD44(200毫秒)和Hoechst特异性信号(6毫秒)的曝光时间保持一致。每个样本的连续切片均使用苏木精和伊红(H&E)染色,并由研究病理学家(M.H.)使用明视显微镜在10倍放大倍率下观察(Olympus IX70)以定位TC和LE/间质区域。
TCGA分析与基因集评分
使用‘UCSCXenaTools’ R包(版本1.4.8)93,94下载了来自国家癌症研究所的癌症基因图谱(TCGA)的患者元数据、生存数据和bulk RNA测序数据。根据患者元数据,从275名患者中筛选出HPV阴性的OSCC样本。对于在至少9/12名患者中差异表达且调整p值 < 0.05的TC和LE基因,使用singscore R包(默认参数)95进行基因集富集分析。核心和前沿基因集评分的最佳分位数切点通过‘survminer’ R包(版本0.4.9)中的‘surv_cutpoint’函数确定,最小比例设为0.2。Kaplan-Meier生存图通过‘survival’ R包(版本3.4-0)中的‘survfit’函数生成,并使用‘survminer’ R包(版本0.4.9)的‘ggsurvplot’函数绘制(https://github.com/kassambara/survminer)。风险比和p值通过‘survival’ R包(版本3.4-0)中的‘coxph’函数进行Cox比例风险测试获得。
在多个癌症类型中比较生存率时,LE基因集评分的最佳分位数切点在每种癌症中均通过‘survminer’ R包(版本0.4.9)中的‘surv_cutpoint’函数确定,最小比例设为0.1。随后使用‘survival’ R包(版本3.4-0)的‘coxph’函数(默认参数)计算优化的风险比和p值。
为进一步评估TC和LE基因集在多个相关临床特征中的表现,作者比较了多种临床特征的有无状态下的TC和LE富集评分,使用双侧Wilcoxon秩和检验,并通过‘stats’ R包(版本4.2.2)应用Benjamini–Hochberg校正以检验显著性。
为检验TC和LE评分之间以及LE评分和CAF评分之间的关系,在数据集中使用‘ggpubr’(版本0.5.0)中的‘stat_cor’函数进行Pearson相关性测试,并通过‘ggscatter’函数绘制。CAF评分使用‘immunedeconv’ R包(版本1.1.5)中的‘EPIC’解卷工具(默认设置)获得96。
TCGA验证
为验证作者的发现,从外部数据库GSE41613下载了包含93名HPV阴性OSCC患者的数据并导入R97。强度数据使用‘limma’ R包(版本3.54.1)98进行预处理和背景校正;基因集通过‘singscore’(版本1.18.0)进行评分,根据优化的切点将其分为高和低TC及LE评分,并计算优化后的风险比和p值。
TCGA分组
通过‘TCGAbiolinks’ R包(版本2.25.3)中的‘PanCancerAtlas_subtypes’函数下载了275个OSCC样本的TCGA亚型数据99。bulk RNA测序数据和亚型数据使用‘CreateSeuratObject’函数添加至Seurat对象(版本4.3.0)99。根据Seurat的映射和注释指南(https://satijalab.org/seurat/articles/integration_mapping.html),在无修改的情况下,将空间转录组位点解卷到HNSC亚型中。亚型数据作为参考数据集,空间转录组数据作为查询数据集。
使用‘singscore’ R包(版本1.18.0)对TC和LE基因集的TCGA亚型数据进行评分。HNSC亚型间的评分差异通过‘ggpubr’(版本0.5.0)中的‘stat_compare_means’函数使用Kruskal-Wallis单因素方差分析进行比较95。
癌细胞状态的机器学习模型
为评估作者在OSCC中识别的核心、过渡和前沿状态是否在其他实体肿瘤中具有保守性,作者使用‘scPred’(版本1.9.2)100训练了三个基于概率的机器学习预测模型(径向基函数核的支持向量机、平均神经网络和朴素贝叶斯)。简要来说,特征选择通过筛选前50个类别信息性主成分(PCs)进行,区分‘核心’、‘过渡’、‘前沿’和‘非癌’的位点级方差。预测模型使用‘caret’ R包(版本6.0-93)进行训练。OSCC数据集中空间肿瘤状态的训练概率使用‘get_scpred’评估,并使用‘plot_probabilities’可视化。径向基函数核的支持向量机用于预测核心、前沿和非癌位点,而朴素贝叶斯用于预测过渡细胞状态。
2例肝胆肝细胞癌(CHC)(Wu等人101),3例肝细胞癌(HCC)(Wu等人101),1例肝内胆管癌(ICC)(Wu等人101),2例皮肤鳞状细胞癌(cSCC)(Ji等人102),1例多形性胶质母细胞瘤(GBM)(10X Genomics),1例人类乳腺浸润性导管癌(HBC-IDC)(10X Genomics),1例人类乳腺浸润性小叶癌(HBC-ILC)(10X Genomics),1例结直肠癌(CRC)(10x Genomics),1例卵巢癌(OV)(10x Genomics),1例肺鳞状细胞癌(10X Genomics),4例皮肤鳞状细胞癌(Abalo等人59),1例前列腺腺癌、浸润性导管癌(10x Genomics),3例胰腺导管腺癌(Lyubetskaya等人103),1例宫颈鳞状细胞癌(10X Genomics),1例前列腺腺泡细胞癌(10x Genomics),1例肠道结直肠癌(10X Genomics),1例黑色素瘤(10X Genomics),1例小儿中枢神经系统胚胎肿瘤(Erickson等人104),以及2例小儿髓母细胞瘤(Erickson等人104),这些样本通过‘scPredict’使用默认概率阈值进行分类和Harmony整合101,102,105。scPred生成的注释被导入‘Seurat’(版本4.3.0)并使用‘SpatialDimPlot’函数叠加在组织图像上。该分类训练模型可通过作者的Figshare门户公开获取(10.6084/m9.figshare.20304456.v1)。
推断细胞通讯网络与分析细胞邻近性
使用‘CellChat’ R包(版本1.6.1)从包含解卷位点的Seurat对象推断细胞-细胞相互作用网络,利用‘createCellChat’函数进行操作106。通过‘netVisual_chord_gene’函数生成过滤后的Circos图以可视化配体-受体对,并基于通讯概率进行阈值调整以优化可视化效果(核心到核心:0.001,前沿到前沿:0.005,CAF和前沿:0.05)。特定通路的Circos图通过‘netVisual_aggregate’函数生成。CellChat还用于在比较模式下通过‘rankNet’函数比较不同信号家族中核心和前沿的总体信息流106。
通过将邻近的非癌位点按最大非癌细胞类型解卷比例进行分类,并统计邻近TC和LE的标注位点的数量来确定非癌位点的绝对数量差异。邻近位点定义为与已标注的恶性TC和LE位点直接接触的位点。这些计数通过双侧Wilcoxon秩和检验比较,并使用‘rstatix’ R包(版本0.7.1)和‘stats’ R包(版本4.2.2)应用Benjamini–Hochberg校正。
构建细胞轨迹的RNA速度分析
为生成推断RNA速度所需的剪接和非剪接数据,使用‘velocyto’命令行界面工具(版本0.17.16)(https://velocyto.org)107。对于每个样本,将对应于病理学标注的癌变区域的条形码和BAM文件输入至velocyto的‘run’命令。‘run’命令提供了由cellranger的‘mkref’函数生成的.gtf基因注释文件,基于标准GRCh38参考基因组生成。输出的loom文件被合并并导入‘scVelo’ Python包(版本0.2.5)中进行进一步分析,降维坐标和scVelo分析的样本元数据从Seurat(版本4.3.0)导入61。多个scVelo对象以相同方式并行处理。
位点级速度基于scVelo的动态模型派生,该模型根据剪接mRNA与未剪接前体mRNA的比率识别位点级轨迹。通过‘scv.pp.moments (data, n_pcs=10, n_neighbors=30)’计算一阶和二阶速度向量矩。动态速度通过‘scv.tl.recover_dynamics’和‘scv.t.velocity’函数计算。基于速度向量的速度置信度使用‘scv.tl.velocity_confidence’函数计算。‘scv.tl.velocity_confidence’函数通过基于相关的方式确定位点级速度向量与其邻近位点间的匹配程度。速度置信度结果通过‘scv.pl.scatter’函数可视化。
使用‘scv.tl.rank_velocity_genes’函数计算每个基因的差异剪接,该函数使用双侧Welch t检验及估算过高的方差来比较多个分组变量并识别组特异性排序基因列表。随后将‘scVelo’的数据导入‘Dynamo’ Python包(版本1.2.0)68,用于向量场学习和位点命运轨迹推断,严格按照其标准操作指南执行。
药物响应与RNA速度轨迹的计算机模拟扰动
通过R包‘PharmacoGx’(版本3.2.0)从CTRPv2、GDSC2、PRISM、CCLE和gCSI数据库下载细胞系药物响应数据108。共识别出五个药物数据集中50种HPV阴性HNSCC细胞系,并找到至少在25种HPV阴性HNSCC细胞系中有可用AAC值的417种药物。对于重复细胞系的数据集,每个细胞系的AAC值在数据集中取平均。每种药物的ACC值通过所有细胞系的AAC值的10%修剪均值计算,以控制药物响应异常值。药物名称随后被传递到药物基因相互作用数据库API(版本2)以识别其下游靶标。不包含上调或下调信息的药物从后续分析中移除。上调由DGIdb关键词“activator”、“agonist”、“inducer”、“partial agonist”、“positive modulator”、“potentiator”和“stimulator”决定;下调由“inhibitor”、“antagonist”、“partial antagonist”、“blocker”、“inverse agonist”、“negative modulator”和“suppressor”决定。最终识别出143种具有下游靶标的药物。如果这些靶标在空间转录组数据集中存在非零表达,这些药物将用于进一步分析。药物靶标被传递至Python包‘Dynamo’(版本1.2.0)以进行计算机模拟扰动分析。通过‘dyn.pd.pertrubation’函数使用Jv缩放因子为−200模拟下调基因,使用因子200模拟上调基因。由扰动产生的速度向量场通过‘dyn.pd.state_graph’函数在‘vf’模式下整合,以估计核心、过渡和前沿状态间的定量位点命运转变概率,并使用‘dyn.pl.state_graph’函数绘制。
‘edge outgoing’特征通过计算‘edge’到‘transitory’和‘edge’到‘core’的位点命运转变概率求和生成。‘core incoming’特征通过计算‘edge’到‘core’和‘transitory’到‘core’的位点命运转变概率求和生成。如果药物的AAC值大于中位数,则分类为‘高’AAC值;小于中位数则分类为‘低’AAC值。位点命运转变概率分数通过‘ggpubr’(版本0.5.0)函数‘stat_compare_means’的双侧配对Wilcoxon秩和检验在‘高’和‘低’AAC组间进行比较。免疫检查点基因CTLA4和CD274分别使用Jv缩放因子为−1000进行计算机模拟扰动。
药物作用机制通过R包‘PharmacoGx’(版本3.2.0)中的PRISM数据库识别。药物名称通过Levenshtein距离算法与PRISM数据集对齐,使用R中的‘Utils’包(版本4.2.2)的‘adist’函数执行,允许字符级差异不超过三的替换、插入、删除或大小写变化。具有相同作用机制的药物被定义为共享机制的药物组(n > 1)。药物类之间的位点命运转变概率通过‘ggpubr’(版本0.5.0)函数‘stat_compare_means’的Kruskal-Wallis单因素方差分析检验进行比较。
空间转录组学图谱
创建了一个网络门户以便于探索所有12个空间样本。该门户通过R包‘shiny’(版本1.7.4)、‘shinyLP’(版本1.1.2)和‘shinythemes’(版本1.2.0)构建,可公开访问,网址为:http://www.pboselab.ca/spatial_OSCC/。用于探索计算机模拟扰动方法的门户也已开放访问,网址为:www.pboselab.ca/dynamo_OSCC。作者的计算机模拟门户还使用了R包‘reticulate’(版本1.2.6)和Python包‘Dynamo’(版本1.2.0)。
统计与重复性
本研究未使用统计方法预先确定样本量。因测序质量不佳,有两个收集的样本被排除在分析之外。实验未进行随机化,研究人员在实验分配和结果评估时未进行盲法处理。
讨论
肿瘤内异质性是癌症患者治疗失败和预后不佳的主要决定因素之一。然而,肿瘤微环境中异质性的空间基础尚不清楚。尽管已认识到肿瘤核心区(TC)和前沿边缘(LE)对肿瘤生物学有深远影响,但这些区域的基因表达特征及其对形成异质性肿瘤微环境的作用尚未被系统性地探索。在此研究中,作者利用12个OSCC组织样本的空间转录组学分析,详细表征了TC和LE的转录组,以更好地揭示它们在OSCC的发生、发展和侵袭中的作用。
首先,作者发现恶性OSCC的TC和LE区域代表了在患者间保守的、具有独特功能特性的不同空间结构。TC呈现角化和分化状态,而LE则赋予肿瘤多种侵袭和转移特性。这些空间结构在预后评估中也具有重要意义——高LE特征分数和低TC分数与患者预后较差相关,而高TC分数则有保护作用,可能部分归因于肿瘤分化水平的增加。有趣的是,这些空间结构在不同癌种中具有通用性。通过作者的机器学习模型,作者发现LE在多种癌症中具有保守性,且可以较高的准确度进行标注,而TC中的基因表达模式可能仅限于相似来源的组织。此外,作者观察到LE特征和TC特征在预后相关性方面的相似趋势:LE特征在多种癌症中具有预后意义,而TC特征的预后价值较为有限,仅在少数癌种中具有显著性。总体而言,作者识别出一个跨癌症类型的保守LE相关转录程序,其与患者预后不良相关。
作者还报告称,TC 和 LE 区域之间的基因表达差异并不受 HNSCC 分子亚型构成或肿瘤进化克隆谱系间的遗传差异的支配。作者的结果表明,一个单一的肿瘤样本中可能同时存在多个 HNSCC 亚型。作者提出,TC 和 LE 的转录组差异是由空间上独特的癌细胞状态驱动的。通过 RNA 速率分析,作者发现这些空间独特的癌细胞状态是由分化层级控制的,即 TC 中具有祖细胞样的癌细胞状态发展为 LE 中更具专业化的癌细胞状态。有趣的是,作者在 TC 和 LE 中的 CSC(癌症干细胞)状态分析中也观察到类似趋势:LE 区域中富含间质样 CSC,而 TC 区域中则有上皮样 CSC。综上所述,作者认为,TC 状态的癌细胞可以通过逐渐获得更具侵袭性的 EMT(上皮-间质转化)样表型,转变为 LE 状态,从而促进癌症的侵袭和扩散。尽管此前已有多项研究描述了转录上独特的癌细胞状态,但作者的研究显示这些状态也受到空间的调控。
最后,作者展示了 RNA 速率在 ST 数据中的一种应用,即预测药物反应。作者发现有效药物(高 AAC 值)通常在逆转 LE 状态的外向转变概率中富集,这突显了 LE 作为一个重要治疗靶点的地位。多种药物可能通过诱导癌细胞向 TC 状态转化来进一步提升疗效。作者假设,有效的抗癌药物能引导癌细胞从 LE 样状态向 TC 样状态转变,从而通过抑制侵袭/转移信号来使这些细胞表型更不具侵袭性。Alvocidib 是一种 CDK 抑制剂,目前正在研究其在急性髓系白血病(AML)中的应用。在作者的分析中,Alvocidib 展现了高于平均水平的 LE 外向和 TC 内向转变信号。此前研究表明,CDK 抑制剂在 OSCC 细胞系中具有良好效果,因此 Alvocidib 可能是进一步研究的有前景的候选药物。重要的是,作者的扰动分析结果表明,空间转录图谱可用于在体外评估药物有效性,并展示了 LE 在肿瘤中作为可信药物靶点的潜力。
作者的研究旨在描述和发现 TC 和 LE 之间基因表达的独特空间模式,并展示了 ST 在 OSCC 中的应用。未来还存在许多令人振奋的研究方向,包括进一步理解癌症侵袭和转移中的保守特征,以及为以 LE 为目标的泛癌治疗方案提供指导。作者相信,本研究获得的时空机制洞见将有助于推动 OSCC 及其他癌症的靶向治疗发展。

汇报人:王肖宇
导师:赵宇
审核:吴婷婷 任建君
