


精读分享│【Nature】:循环代谢生物标志物的全基因组特征分析
英文题目:Genome-wide characterization of circulating metabolic biomarker
中文题目:循环代谢生物标志物的全基因组表征
期刊:Nature(IF: 64.8)
单位:芬兰奥卢大学医学院系统流行病学系及奥卢生物中心等
发表时间:2024年4月


背景:
该研究使用高通量代谢组学平台进行的全基因组关联分析为人类代谢生物学提供了新的见解。对全身代谢的遗传决定因素的详细了解对于揭示遗传途径如何影响生物机制和复杂疾病至关重要。 本文介绍了一项针对233个循环代谢性状的全基因组关联研究,这些性状是通过核磁共振光谱法在来自33个队列的136,016名参与者中定量的。该研究鉴定了400多个独立的基因座,并手动整理了潜在的生物学候选基因,在其中三分之二的基因座上分配了可能的因果基因。 该研究强调了样本和参与者特征的重要性,这些特征可能对遗传关联可能产生显著影响。该研究利用脂蛋白和脂质相关变异的详细代谢特征,更好地表征了已知的脂质基因座和新发现的基因座对脂蛋白代谢的影响。此外,该研究展示了全面表征的分子数据的转化应用,描述了妊娠期肝内胆汁淤积的代谢关联。 最后,该研究观察到多条代谢途径的显著遗传多效性,说明了在孟德尔随机化分析中仔细选择工具的重要性,并揭示了丙酮与高血压之间可能的因果关系。本文的公开结果为研究代谢在不同疾病中的作用提供了基础资源。 方法: 1、NMR代谢组学 在这项工作中,该研究扩展了之前的大约25,000人中的123种人类代谢特征的全基因组关联研究(GWAS),包括了额外的队列和更全面的代谢特征面板。在33个队列中(总样本量高达136,016)使用与之前研究相同的NMR代谢组学平台的更新版本对最多233种血清或血浆代谢特征进行了量化。NMR代谢组学平台提供了脂蛋白亚类及其脂质浓度和组成、apoAI和apoB、胆固醇和甘油三酯测量、白蛋白、各种脂肪酸和低分子量代谢物的数据,例如氨基酸、糖酵解相关测量和酮体。在本研究中,代谢特征在以下队列中被量化:埃文儿童与家长纵向研究(ALSPAC)、中国科学院(CKB)、塔尔图大学爱沙尼亚基因中心队列(EGCUT)、鹿特丹欧拉斯家庭研究(ERF)、欧洲遗传数据库(EUGENDA)、FINRISK 1997(FR97)、FINRISK 2007(FR07,即DILGOM)、间隔生物资源(INTERVAL)、克罗地亚科尔丘拉研究(KORCULA)、LifeLines-DEEP(LLD)、莱顿长寿研究(LLS)、伦敦生命科学前瞻性人群研究的八个亚队列(LOLIPOP)、男性代谢综合征研究(METSIM)、荷兰肥胖流行病学研究(NEO)、荷兰抑郁和焦虑研究(NESDA)、1966年北芬兰出生队列(NFBC1966)、1986年北芬兰出生队列(NFBC1986)、荷兰双生子登记(NTR)、牛津生物银行(OBB)、奥克尼复杂疾病研究(ORCADES)、普拉瓦斯坦老年人风险前瞻性研究(PROSPER)、鹿特丹研究的三个亚队列(RS)、TwinsUK(TUK)、年轻芬兰人的心血管风险研究(YFS)。大多数队列由欧洲血统个体组成(6个芬兰和21个非芬兰),六个队列由亚洲血统个体组成(1个汉族中国人和5个南亚人)。 2、全基因组关联研究 在33个队列中对233种代谢特征(补充表2)进行了GWAS(补充表1),最多包括136,016名个体,这些个体拥有NMR代谢特征测量和全基因组SNP数据。研究中排除了怀孕的个体或那些使用降脂药物的个体。通过使用单倍型参照联盟(Haplotype Reference Consortium)版本1.1或1000基因组项目第三阶段版本进行SNP推断,并且在每个队列中分别按加性模型进行了GWAS。在分析前,代谢特征分布根据年龄、性别、主成分及相关的研究特定协变量进行了调整,并对特征残差进行了逆秩正态转换。 队列在固定效应meta分析中结合使用了METAL软件,且仅过滤出至少出现在七个队列中的SNP。NMR代谢特征高度相关,使用Bonferroni校正来考虑多重检验将导致全基因组显著性阈值过于保守。因此,该研究使用INTERVAL中定义的主成分数量(28个)解释代谢特征中>95%的变异来校正多重检验,该研究的全基因组显著性阈值设为p<1.8×10-9(标准全基因组显著性水平p<5×10-8,除以28)。在初步GWAS之后,对233种代谢特征进行了禁食和样本类型分层分析。在这些分析中,26个队列被归类为禁食(n=68,559),6个队列归类为非禁食(n=58,112),17个队列归类为使用血清样本(n=90,223)和16个队列使用血浆样本(n=45,793;见补充表1)。为了定义跨代谢特征的相关位点,该研究定义了每个达到显著性阈值的SNP两侧的500 kb窗口,将所有代谢特征的这些窗口在每条染色体上合并,然后迭代合并窗口。由于这种方法可能导致多个独立信号被包括在这些位点内,该研究进一步根据成对连锁不平衡(LD,间隔和FINRISK97中定义的r2截止值为0.3)定义了位于已定义位点内的潜在独立信号。在LocusZoom(v.1.4)中创建了区域关联图。该研究根据两个标准指定了相关的lead SNP到最可能的因果基因:(1)该研究优先考虑与相关代谢特征具有明确生物学相关性的基因;及(2)如果没有检测到生物学上可信的因果基因,且lead snp是功能性变异(错义、剪接区域或获得终止)或与此类变异高连锁不平衡(INTERVAL中的r^2 >0.8),则带有功能变异的基因被指定为最可能的候选基因。(3)如果未满足标准1和2,则最近的基因被指示为候选基因。 3、特定祖先的分析 该研究在主要的发现性meta分析中进行了祖先分层分析,包括南亚人(5个队列,11,340名参与者)、东亚人(1个队列,4,435名参与者)、所有欧洲人(27个队列,120,241名参与者)、芬兰人(6个队列,27,577名参与者)和非芬兰欧洲人(21个队列,92,664名参与者)。对于这些特定祖先的分析,该研究使用了全基因组显著性的标准阈值(p<5×10-8)。为了与具有非洲祖先的参与者进行比较,该研究使用了UK Biobank数据集(2021年3月发布)。根据基线问卷中的自我报告的种族信息(字段21000:种族背景),该研究确定了具有加勒比(代码4001)、非洲(代码4002)或任何其他黑人背景(代码4003)的非洲祖先的1,405名参与者。变异的质量控制通过排除最小等位基因频率<1%、INFO得分<0.4的SNP以及复杂LD区域中的变异进行。LD thinning(是一种用于遗传关联研究(GWAS)中的数据处理方法,通过从数据集中删除高度相关的SNP来减少LD的方法。通常,这是通过设置一个LD阈值来实现的,例如r²(相关系数的平方)小于某个给定值)使用r2<0.1,窗口大小为1,000,步长为80进行。通过UK Biobank提供的相关性数据(字段22021:与其他参与者的遗传亲缘关系)确定并排除相关个体。从非相关样本中计算的前6个遗传主成分的异常值被从分析中删除。NMR代谢特征根据年龄、性别、禁食状态和10个遗传主成分进行调整,并且特征残差被逆秩正态转换。使用PLINK 2.0测试SNP与代谢特征之间的关联。 4、在公开可用数据中的复制 从IEU Open GWAS项目下载了UK Biobank SNP-代谢特征的汇总统计数据(https://gwas.mrcieu.ac.uk/datasets/?gwas_id__icontains=met-d)。这些汇总统计数据来源于2021年3月公开发布的UK Biobank数据,其中的代谢特征是用类似于该研究中使用的新版夜莺健康(Nightingale Health)平台的NMR技术测量的。这些数据用于比较该研究的lead snp-代谢特征对在276个相关区域内的关联。在UK Biobank数据中定义关联使用了两个阈值:标准的全基因组显著性水平(p<5×10-8)和提示性显著性水平(p<1×10-5)。 5、遗传力和方差解释 该研究使用GCTA-GREML v. 1.94估计了每个特征的常见变异的遗传力,使用的是独立的数据集,具体是UK Biobank第一阶段NMR发布的数据。这项研究是在UKBB资源下的申请编号7439进行的。该研究随机选取了10,000名具有可用NMR数据且不相关的欧洲血统UK Biobank参与者,并将推断变异筛选到次等位基因频率>0.005,缺失率<0.1,以及哈代-温伯格平衡P值<10^-6。该研究使用之前描述的方法消除了特征的技术变异,并根据年龄、性别、降脂药物使用和前10个遗传主成分调整了特征。在GREML分析之前,特征进行了逆秩正态转换。每个特征的lead snp解释的方差如之前描述的那样进行了估计。 6、与之前关联的比较 该研究对代谢特征关联进行了广泛的比较,与之前的代谢特征GWAS研究相对照。该研究分为三组:(1)与之前发布的大型循环NMR特征的GWAS结果比较;(2)与临床脂质相关的位点比较(包括来自UK Biobank 2019年9月版本3的数据);(3)与之前的代谢物和代谢组学研究的广泛关联列表比较。比较是通过指出:(1)共定位的已知变异;(2)lead snp 500 kb侧翼内的任何已知关联;或(3)与lead snp处于连锁不平衡(LD,r2 > 0.3,在INTERVAL中定义)的已知关联来进行的。由于该研究使用了UK Biobank进行复制,没有将关联与那些使用UK Biobank NMR代谢组学作为单一队列而没有验证队列的研究进行比较。 除了与之前的代谢特征关联进行比较外,该研究还使用PhenoScanner(v2.44,45)和NHGRI-EBI GWAS目录(2023年3月30日使用gwasrapidd R包版本0.99.1489下载的关联数据)筛选了lead snp的先前疾病和特征关联(P值截止值5×10-8)。此外,该研究还筛选了FinnGen数据的3,095个疾病终点的汇总统计数据,寻找重叠关联(P值截止值5×10-8)。使用PhenoScanner(v2.44,45)筛选与基因表达和蛋白水平的关联。 7、脂蛋白基因座的代谢作用 为比较脂蛋白、脂质和载脂蛋白相关变异的代谢效应,效应估计值以颜色编码的热图形式呈现。为了允许比较SNP效应,将估计值相对于每个SNP的最高绝对值进行了缩放。在此分析中,该研究包括了276个最初定义的区域中的lead snp,这些SNP在全基因组显著性水平上与任何脂蛋白脂质或载脂蛋白相关,并在名义上(P<0.05)与apoB相关。该研究使用这些标准限制分析至与apoB相关的SNP,因为已知apoB是心血管疾病的脂蛋白代谢的因果部分。为排除由于相同因果基因导致的在代谢特征上具有相似效应的信号,如果代谢特征关联的模式相似(R>0.5),该研究只从最初定义的具有多个独立信号的基因组区域中包括单个SNP。 在热图中,每一行代表一个单独的SNP,每一列对应一个单独的代谢测量,SNP-代谢物关联的缩放效应估计值通过颜色范围进行可视化。效应的方向显示与增加apoB相关的等位基因的关系。为了将具有相似效应的SNP分组,基于缩放SNP效应的层次聚类构建了树状图。使用gplots v. 3.0.3 R包中的heatmap.2函数构建热图。在R v. 4.0.0中评估了Pearson相关性。 8、妊娠期肝内胆汁淤积症(ICP) 该研究使用FinnGen研究的数据(O15_ICP;1,460例病例,172,286名对照)的汇总统计数据评估了其代谢特征关联与ICP的重叠。ICP病例通过医院出院登记定义,使用ICD10代码O26.6以及ICD9代码6467A和6467X。使用每个相关位点的最近基因,进行了基因本体(GO)富集分析,以搜索富集的生物过程和分子功能GO术语。该研究使用多特征共定位的假设优先级排序(HyPrColoc)R库(版本1.0)评估关联信号的共定位,在该库中使用高效的确定性贝叶斯算法同时检测大量特征的共定位。寻找单一因果变异和共享区域关联的共定位。 为了可视化SNP在脂质和脂蛋白特征中的效应,使用gplots v. 3.0.3 R包的heatmap.2函数构建了热图。热图中包括以下SNP:GCKR-rs1260326、ABCB11-rs10184673、ABCB1-rs17209837、CYP7A1-rs9297994、SERPINA1-rs28929474和HNF4A-rs1800961。代谢特征相关SNP的效应相对于ICP的1.5的比值进行了缩放。 9、孟德尔随机化分析 使用来自UK Biobank的20个NMR非脂质代谢特征(包括氨基酸(丙氨酸、谷氨酰胺、甘氨酸、组氨酸、异亮氨酸、亮氨酸、缬氨酸、苯丙氨酸和酪氨酸)、酮体(乙酸盐、乙酮和3-羟基丁酸)以及糖酵解/糖异生(葡萄糖、乳酸、丙酮酸、甘油和柠檬酸)、液体平衡(白蛋白和肌酐)或与炎症相关(糖蛋白乙酰化)的代谢特征)作为暴露因子,以及UK Biobank的460个Phecodes和52个定量特征作为结果变量进行了双样本孟德尔随机化分析。 该研究为分析定义了两组工具,称为全面和严格的工具。作为初步工具,使用了与这些特征相关的334个lead snp(每个定义的相关位点一个工具SNP)(“全面工具”)。为避免由多效性引起的潜在偏差,该研究还选取了193个变异的子集(“严格工具”),这些变异在所有233个代谢特征中的关联数少于5。该研究基于经验评估每个变异的特征关联分布来确定5个关联的阈值。 为了探究孟德尔随机化分析对阈值选择的敏感性,该研究还测试了关联数少于3个和少于7个的情况。使用PheWAS R软件包中提供的主要Phecodes精选列表在UK Biobank中定义疾病结果。为了将分析限制在主要疾病结果上,该研究丢弃了任何子类别(即,Phecodes具有4个或更多字符)并移除了在多达367,542名不相关的欧洲血统UK Biobank参与者中发病次数少于100的结果。最终得到的460种疾病被分为15个广泛的领域:循环系统、皮肤科、消化系统、内分泌/代谢、泌尿生殖系统、造血系统、传染病、精神障碍、肌肉骨骼系统、肿瘤、神经系统、妊娠并发症、呼吸系统、感官器官和症状。该研究还分析了UK Biobank中提供的52个定量特征,包括血压、肺功能测量、血细胞特征和临床化学生物标志物。在该研究的复制分析中(以乙酮作为暴露因子和高血压作为结果),使用了FinnGen研究数据中的必需高血压作为结果(高血压本质,I9_HYPTENSESS;70,651例病例,223,663名对照)。病例通过医院出院登记定义,使用ICD10代码I10,ICD9代码4019X和4039A,ICD8代码40199、40299、40399、40499、40209、40100、40291、40191和40290。 该研究针对每个工具使用了逆方差加权方法进行了单变量孟德尔随机化分析,还进行了敏感性分析,使用MR-Egger回归来考虑未测定的多效性,并使用Weighted Median回归评估对无效遗传工具的鲁棒性。该研究的主要分析基于固定效应模型,但作为敏感性分析,使用了随机效应模型来考虑变异间的异质性,使用I-squared统计量来量化。孟德尔随机化分析使用MendelianRandomization软件包v.0.5.1或TwoSampleMR软件包v.0.5.3进行。单SNP孟德尔随机化估计基于Wald比率。该研究认为固定效应的逆方差加权方法是主要的孟德尔随机化模型。为了考虑多重检验,认为P值小于4.88 × 10−6的关联是显著的(Bonferroni校正,用于对20个代谢特征进行512个结果的测试)。 10、芬兰基因研究(FinnGen) 该研究中使用了来自FinnGen数据的3,095个疾病终点的GWAS汇总统计数据。FinnGen研究的完整描述和数据分析步骤提供在补充说明中。FinnGen的贡献者列在补充表18中。 统计与可重复性 meta分析由两个不同中心的两位研究者独立进行(芬兰奥卢大学和英国剑桥大学),并比较了汇总统计数据以验证结果的一致性。 结果: 1、遗传发现 在33个队列中分别采用加性模型进行了GWAS(补充表1)。后续的meta分析涉及233种代谢特征(补充表2),包括213种脂质和脂蛋白参数或脂肪酸以及20种非脂质特征(氨基酸、酮体和糖酵解/糖异生、液体平衡和炎症相关代谢物)。在变异过滤和质量控制后,多达13,389,637个模拟的常染色体单核苷酸多态性(SNPs)被纳入最多136,016名参与者的meta分析中。
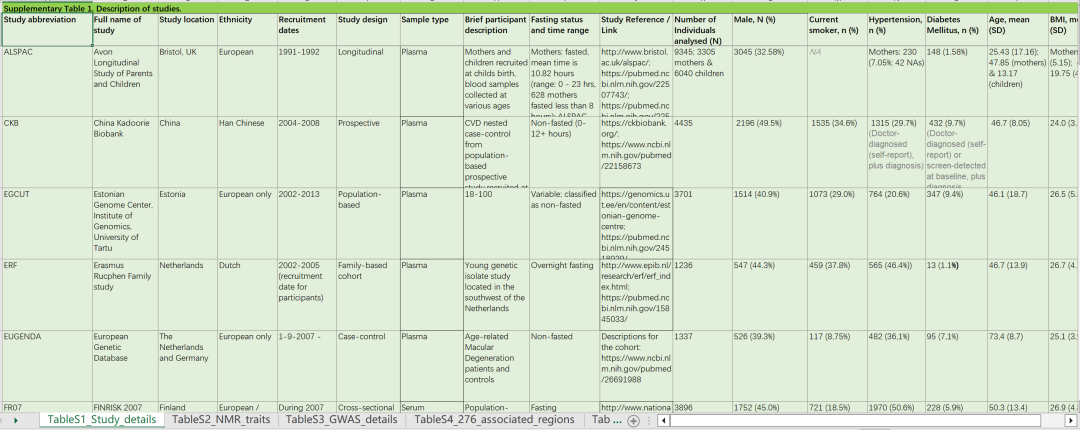
补充表1 | 研究队列信息
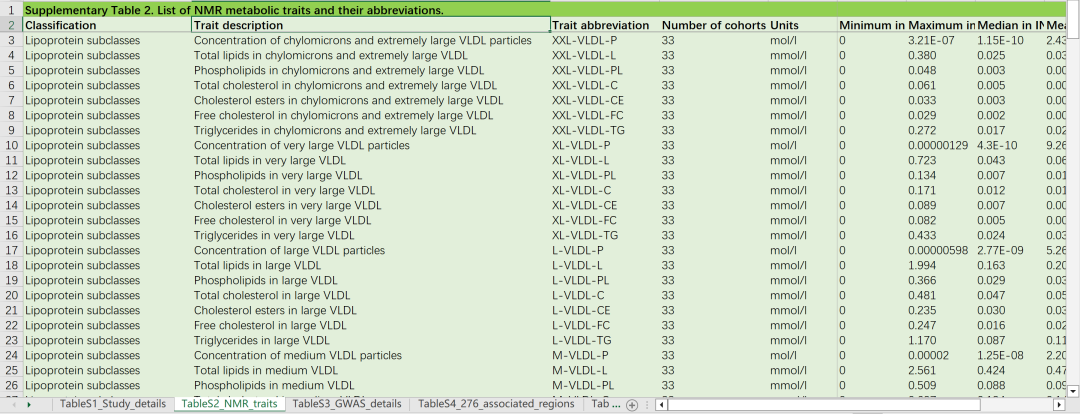
补充表2 | NMR代谢特征信息
在meta分析中,该研究检测到了所有233种代谢特征的全基因组显著关联,并发现了广泛的多效性和多基因性。检测到了276个广泛区域(定义为全基因组显著SNPs周围±500kb的区域)与至少一种代谢特征相关(图1a)。其中86个区域仅与单一代谢特征相关,而大多数区域则与多种特征相关(图1b,c),最多达214种特征与已充分表征的脂质相关APOE区域相关。 脂质、脂蛋白和脂肪酸特征主要表现出多基因性,有60种特征在超过50个基因座上存在关联,137种特征(64.3%)在20—50个基因座上存在关联,16种特征(7.5%)在少于20个基因座上存在关联。 大多数非脂质特征的相关基因座显著较少(13种特征少于20个相关基因座;所有20种非脂质特征中的65%),包括3种葡萄糖代谢相关特征(乳酸、丙酮酸和甘油)有少于5个相关基因座,而糖蛋白A和一些氨基酸在20—33个基因座上存在关联,肌酸在49个基因座上存在关联。非脂质特征占据了大多数单一相关特征区域(n = 67;78%),并且大多数(n = 163;57%)与非脂质特征相关的区域有总共少于5个相关代谢特征。相比之下,脂质、脂蛋白和脂肪酸特征相关区域(n = 186)通常表现出更多的多效性,其中75%(n = 140)的区域与5种或更多特征相关。这种多效性差异是由于脂蛋白代谢是一个连续体,基因往往影响多个颗粒类别,而非脂质特征通常受更为不同的过程和酶修饰的影响,从而导致较少的多效性。在276个区域内,发现了8,795个lead snp-主要特征关联,涉及1,447个独特的lead snp。在基于成对连锁不平衡(LD)解析独立信号后,得出这276个广泛区域涉及至少443个独立基因座。 该研究估计了在UK Biobank中也有记录的223种特征的全基因组常见变异遗传率。中位特征遗传率为0.29,其中仅约四分之一由lead snp解释,支持了许多特征的高度多基因性。
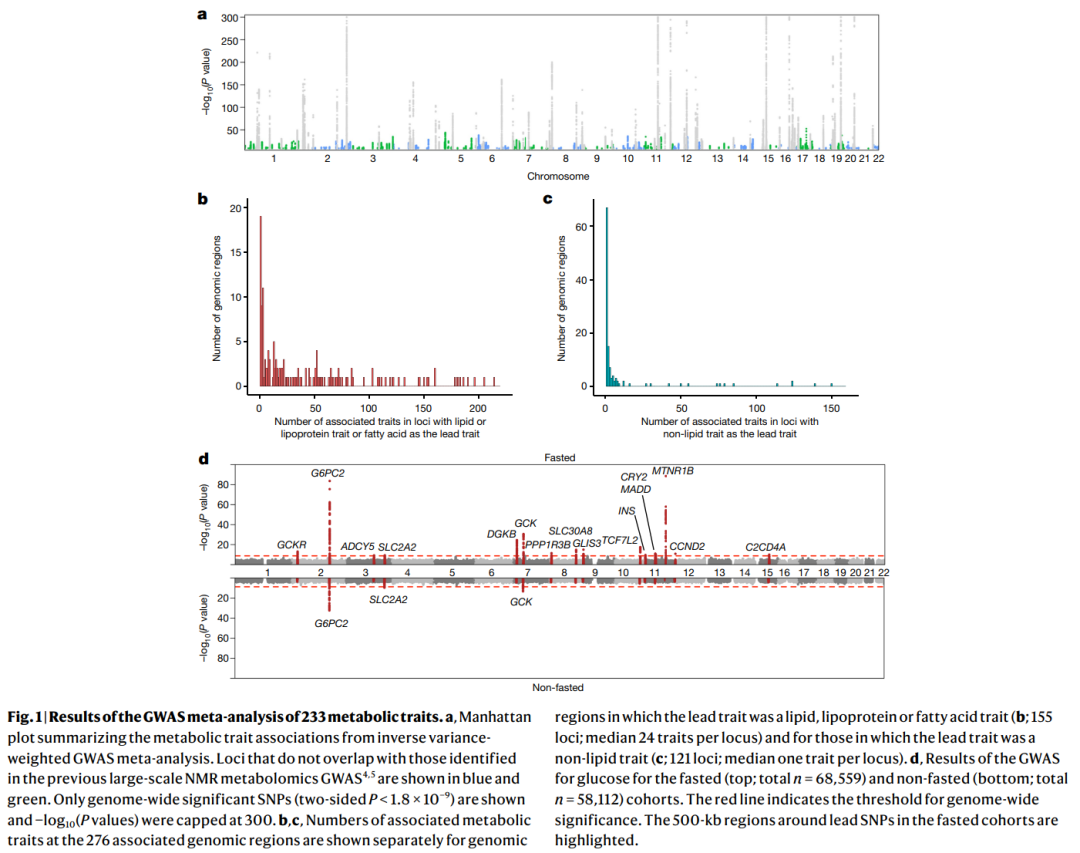
图1 | 233种代谢特征的GWAS meta分析结果。
a. 曼哈顿图总结了逆方差加权的GWAS meta分析中的代谢特征关联基因座。蓝色和绿色显示不与先前大规模NMR代谢组学GWAS中识别的区域重叠的基因座。仅显示具有全基因组显著性SNP(双侧P值<1.8×10−9),并且-log10(P值)被截断为300。 b,c. 分别显示了在基因组区域中关联的代谢特征的数量,其中首要特征是脂质、脂蛋白或脂肪酸特征(b; 155个基因座;每个基因座的中位数为24个特征)和首要特征为非脂质特征(c; 121个基因座;每个基因座的中位数为一个特征)。 d. 禁食(顶部;总n=68,559)和非禁食(底部;总n=58,112)群体中葡萄糖的GWAS结果。红线表示全基因组显著性的阈值。突出显示了群体中lead SNP周围的500 kb区域。 2、祖先分层分析 为了探究该研究的关联结果在不同族群间的普适性,并寻找额外的祖先特定的关联信号,对南亚人(五个队列,11,340名参与者)、东亚人(一个队列,4,435名参与者)、芬兰人(六个队列,27,577名参与者)和非芬兰欧洲人(21个队列,92,664名参与者)进行了祖先分层分析(补充表7)。为了进一步探究普适性,该研究额外对具有非洲祖先的小型人群(n=1,405)的关联进行了事后比较。关联在不同祖先群体间强烈正相关(扩展数据图1),表明关联结果在不同祖先之间具有广泛的可传递性。芬兰人和非芬兰欧洲人之间的相关性更强(r=0.96),与东亚人和南亚人(r≈0.7)以及非洲人(r≈0.4)相比。对于一些基因座,效应估计在一个祖先中明显比另一个更强。全基因组显著关联的数量与样本量强相关,从非芬兰欧洲人中的7,002个关联到东亚参与者的331个和非洲参与者的97个不等。在任何祖先群体中都没有检测到新的基因组区域,超出了在祖先组合的meta分析中发现的276个,这表明在未来的研究中,需要更多具有非欧洲祖先的参与者的样本。
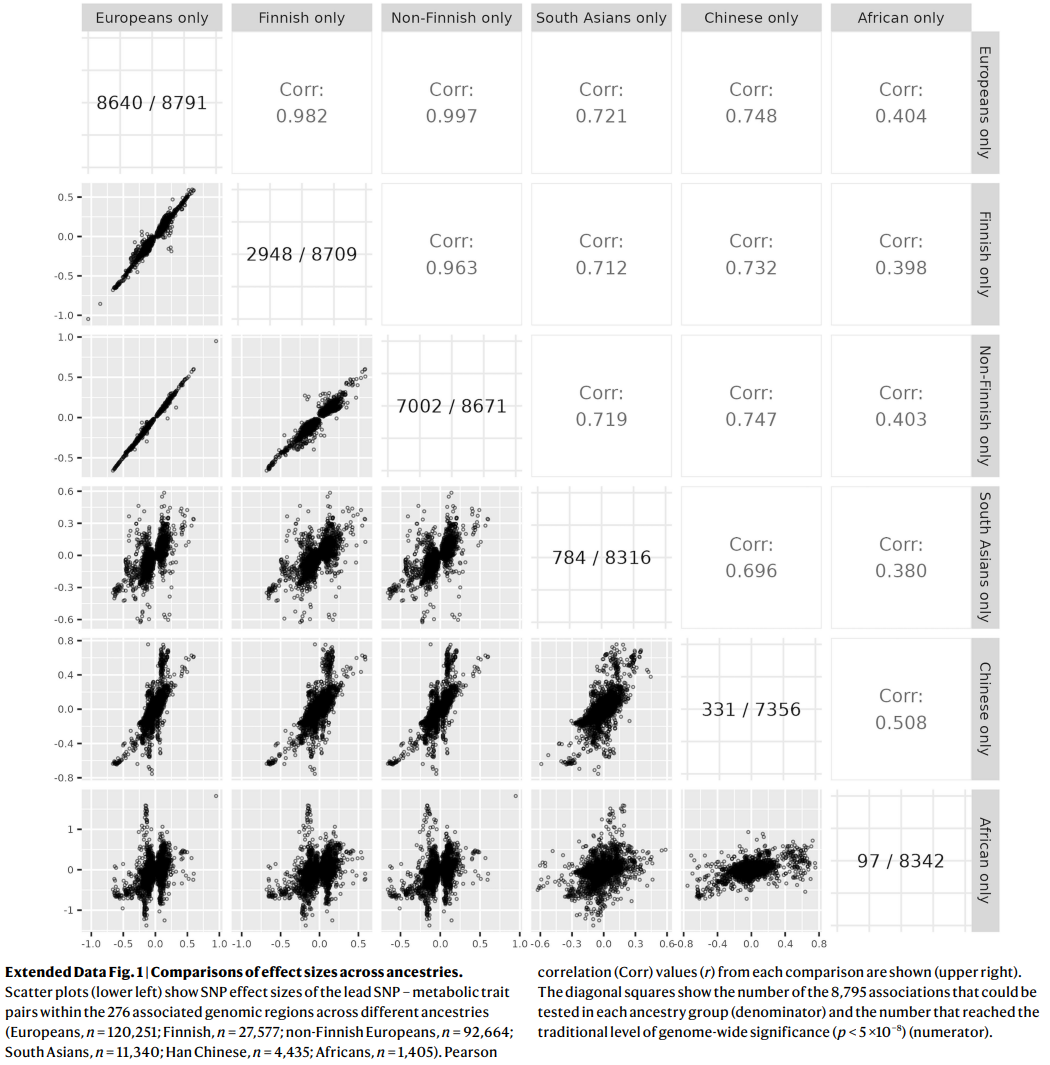
扩展数据图1 | 不同人种间效应大小的比较。
散点图(左下角)显示了跨越276个相关基因组区域的首要SNP—代谢性状对在不同人种间的SNP效应大小(欧洲人,n=120,251;芬兰人,n=27,577;非芬兰欧洲人,n=92,664;南亚人,n=11,340;汉族,n=4,435;非洲人,n=1,405)。每次比较的皮尔逊相关系数(Corr)值(r)显示在右上方。对角线方块显示了在每个人种群中可以测试的8,795个关联性(分母)以及达到传统全基因组显著水平(p<5×10−8)的数量。 3、在UK Biobank中的关联 UK Biobank资源的NMR数据(2021年3月发布)使该研究能够在一个独立的人群中检查主要变异体的关联,并评估参与者特征和样本相关因素对其关联的影响。在多达115,078名欧洲祖先的UK Biobank参与者中,有8,502个lead snp-代谢特征对可以进行测试,其中5,442个(64.0%)在p<5×10−8的水平上关联,并有进一步的772个(9.1%;328个唯一SNPs)在p<1×10−5的水平上关联。当在具有不同样本类型(血清,n=90,223;血浆,n=45,793)以及禁食(n=68,559)和非禁食(n=58,112)的队列中进行进一步分层分析时发现,除了研究之间人群祖先的细微差异外,样本类型和禁食状态可能是不相同的主要因素。UK Biobank的NMR测量是在EDTA血浆样本上进行的,而当前的meta分析主要涉及血清样本。例如,几个与苯丙氨酸无关的关联是在与凝血相关的基因座上(例如KLKB1、F12、KNG1和FGB)发现的,但这些信号在UK Biobank中不存在- 因此他们推测在制备血清时去除凝血因子可能会揭示出与苯丙氨酸的关联。两个基因座(NHLRC1,lead snp rs73726535;TXNRD1,lead snp rs191631370)在UK Biobank中也与苯丙氨酸相关,但在当前的meta分析中不存在。类似地,该研究发现了与葡萄糖相关的关联,在UK Biobank中无法复制,包括在褪黑激素受体1B基因22(MTNR1B)中的一个众所周知的关联,这是葡萄糖代谢中的关键调节因子(rs10830963;meta分析P值= 1.5 × 10-60;UK Biobank P值= 0.60)。UK Biobank主要包括非禁食样本,但当前的meta分析主要由禁食样本组成(26个队列)(补充表1),该研究的禁食分层meta分析表明,一些葡萄糖相关的关联是由主要是禁食样本的队列推动的(图1d),因此在UK Biobank中不存在。除了MTNR1B rs10830963(在禁食和非禁食队列的meta分析中的P值分别为2.9×10−89和0.57),该关联先前也被证明在非禁食样本中不存在23,GLIS家族锌指3(GLIS3;已知的与糖尿病风险相关的基因24,在胰岛β细胞生物学中发挥作用)rs10974438代表了另一个在UK Biobank中没有得到稳固复制的关联的示例(meta分析P值= 4.0×10−14;UK Biobank P值=0.001),并且在非禁食队列中不存在信号(禁食和非禁食队列的meta分析P值分别为1.1×10−15和0.14)。 许多代谢特征关联受到样本类型和禁食状态的影响,尽管与总体关联的比较因分层分析的功效降低而复杂化。例如,几种脂蛋白亚类的关联在涉及脂类生物学核心角色的位点(如LPA和ANGPTL3)受禁食状态显著影响。类似地,多个位点在使用血清的队列中相比使用血浆的队列有超过两倍的效应估计高或低。这些差异不仅在脂质特征中检测到,也在非脂质特征中检测到,一些关联通过去除血浆样本显著增强。然而,在按样本类型分层的分析中仅检测到7个额外的位点(超出最初关联的276个基因组区域),而在禁食分层分析中检测到10个(补充表10和11)——例如,仅在禁食队列中显示关联的C2CD4A rs10083587对葡萄糖和KAT5 rs12787843对肌酐(P值分别为1.3×10−10和7.6×10−10)。他们注意到,在解释代谢特征GWAS的结果和进行下游分析时,如使用与特征相关的变异体作为工具的孟德尔随机化研究,需要仔细考虑样本类型和禁食状态的影响。 4、新发现的位点和候选基因 作者进行了广泛的手动整理,以优先考虑297个位点中的231个具有明确生物学相关性的可能因果基因(占443个位点的67.0%)。由于某些区域极其复杂且多效性,这是由于多达11个独立的主要变异体与代谢特征间存在重叠的遗传关联,详细描述了这些位点,以确定每个位点内可能的多个潜在因果基因。例如,在16号染色体上一个7.6兆碱基(Mb)的区域内,有139个相关的代谢特征,鉴定出6个不同的生物学相关的潜在因果基因:卵磷脂-胆固醇酰基转移酶(LCAT;与多种脂蛋白亚类测量相关)、溶质载体家族7成员6(SLC7A6;与乙酸和肌酐相关)、丙酮酸脱氢酶磷酸酶调节亚单位(PDPR;与丙酮酸和氨基酸相关)、丙氨酰-tRNA合成酶1(AARS;与丙酮酸和氨基酸相关)、酪氨酸氨基转移酶(TAT;与酪氨酸相关)和血红蛋白(HP;与多种脂蛋白亚类测量、脂肪酸、胆固醇、载脂蛋白B(apoB)和糖蛋白乙酰化相关)。此位点体现了与代谢特征相关的位点的复杂性。 对于没有明显生物学候选者的额外位点,作者根据SNP功能或与主要变异体强连锁不平衡(LD,r2 ≥ 0.8)的可能功能性(错义、获得终止或剪接区域)变异,进一步指派了39个可能的因果基因(补充表5)。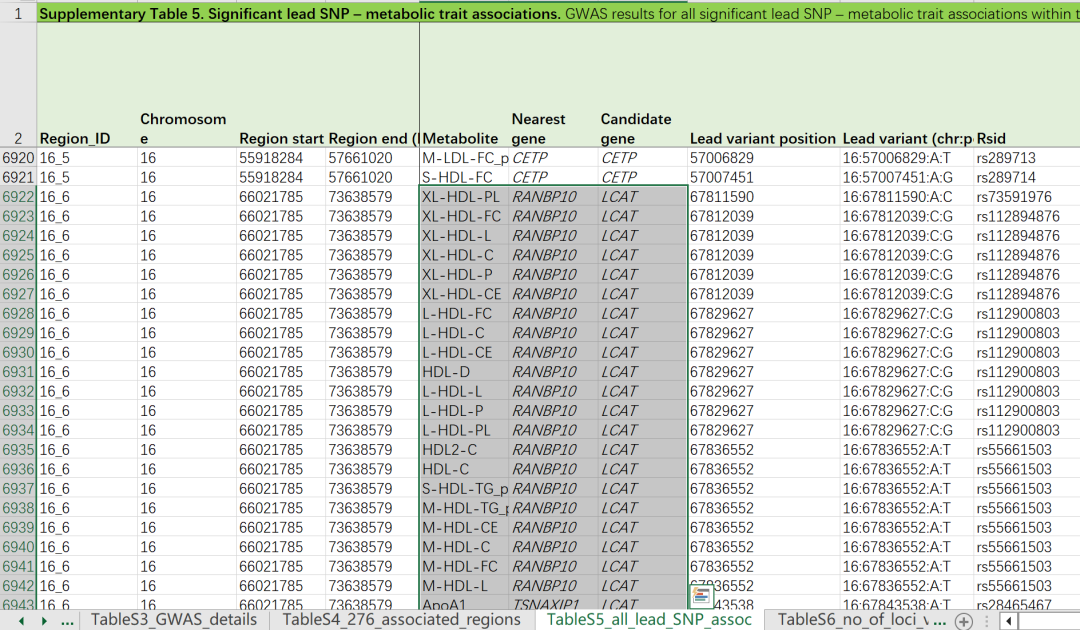
补充表5
作者对发现的关联进行了广泛比较,以前报道的代谢特征和传统临床脂质(高密度脂蛋白C(HDL-C)、低密度脂蛋白C(LDL-C)、甘油三酯和总胆固醇)的遗传关联进行了对比(补充表5)。与先前的大规模NMR代谢组学GWAS相比,确定了212个额外的关联基因区域。其中包括138个新的基因组区域与脂蛋白、脂质和脂肪酸特征相关,以及113个新的与非脂类特征相关的区域。在以前与临床脂质相关的位点中,检测到了多个脂蛋白亚类测量相关的新关联,如含有低密度脂蛋白受体适配蛋白1基因(LDLRAP1)的位点,该基因参与胆固醇代谢。此位点先前已知与LDL-C、甘油三酯和总胆固醇相关,在该位点发现了与多种脂蛋白亚类测量、脂质和脂肪酸相关的关联(补充表5)。含有甾醇O-酰基转移酶2基因(SOAT2)的位点(在胆固醇代谢中发挥作用)是另一个新的与脂蛋白和脂质特征相关的生物学合理位点的例子。作者的分析还确定了在以前未报告与传统临床脂质相关的位点中与详细的脂蛋白亚类测量相关的遗传关联。与迄今为止最大的跨族群临床脂质研究相比,检测到了十二个额外的关联位点(补充表5),涉及脂蛋白和脂质特征(对应所有脂蛋白和脂质特征相关区域的6.5%):编码2型乳糖氨酸α-2,3-唾液酰基转移酶(ST3GAL6)的基因是一个与多个脂蛋白亚类测量和脂质相关的生物学合理基因的例子。 作者还检测到了与苯丙氨酸和谷氨酰胺等小分子相关的新位点。对于苯丙氨酸,检测到了13个位点的关联。新的苯丙氨酸相关位点包括一个众所周知的代谢特征相关位点(FADS1–FADS2)和两个新的、生物学上合理的位点(GSTA2和SLC2A4RG)。例如,SLC2A4RG编码SLC2A4调节因子,这是一个涉及溶质载体家族2成员4(SLC2A4,也称为GLUT4)的激活的转录因子,它是葡萄糖转运的关键调节因子。对于谷氨酰胺,检测到了26个位点的关联。值得注意的是,其中有七个位点仅与谷氨酰胺相关(GLS、PLCL1、SFXN1、KCNK16、MED23、SLC25A29和PCK1)。因此,这些关联很可能代表了与谷氨酰胺有关的生物学,其中大多数位点都有生物学上合理的候选基因,这些基因在谷氨酰胺代谢(GLS)、氨基酸运输(SFXN1和SLC25A29)或葡萄糖和糖异生途径(PCK1和KCNK16)中发挥作用。KCNK16是一个已知的2型糖尿病易感基因,编码钾通道亚家族K成员16,是胰岛钾通道的一个例子,代表了一个在葡萄糖生物学中发挥作用的新的与谷氨酰胺相关的位点。 5、载脂蛋白B(apoB)变异的影响 为了提供脂质位点如何影响脂蛋白代谢连续体的不同方式的见解,作者对具有相似代谢关联特征的基因群进行了表征。效应估计通过用给定SNP的最强关联效应估计值除以所有效应估计值来进行缩放。这样,所有SNP的缩放效应估计值都在-1到1之间,关联的统计强度对聚类的影响较小,更多的重点放在关联景观上来指导聚类。该研究专注于有明显证据(p<0.05)与载脂蛋白B(apoB)相关的134个位点,因为最近的研究强调了apoB在冠状动脉疾病病因中的主导作用。这些位点的聚类产生了至少六个主要的位点群(扩展数据图4和5)。扩展数据图4中的最上方的群与以前观察到的与2型糖尿病风险和肥胖相关的流行病学关联特征非常相似。上方第二个群(扩展数据图4和5)主要显示三酰甘油富集的非常低密度脂蛋白(VLDL)颗粒增多和大型HDL颗粒减少。这个聚类中的基因,如LPL、MLXIPL和ANGPTL4,与三酰甘油代谢和葡萄糖代谢相关,例如GCK、GCKR和INSR所示。其他聚类主要与LDL颗粒相关,与其他脂蛋白关系较少。最下方的群包括在血液中影响LDL胆固醇的生物学相关基因,包括APOB、LDLR、PCSK9、SORT1和HMGCR。尽管脂质和载脂蛋白特征内部的强相关结构,作者还是发现了几个与特征间的相关结构不符的关联模式的位点(图2a和扩展数据图4和5)。例如,一些位点(APOC1和TIMD4)强烈与所有含有apoB的颗粒(VLDL、中密度脂蛋白(IDL)和LDL)相关,而其他位点主要与IDL和LDL颗粒相关(PCSK9、HMGCR和TRIM5),或与VLDL和最大的HDL颗粒(IRS1和CD300LG),或与中等和小HDL颗粒(APOA2和CERS2)相关。一些SNP也在高度相关的代谢特征内显示出不一致的关联(例如,LPA和APOH SNP在含有apoB的颗粒内,以及FADS簇SNP在含有apoB和HDL的颗粒内;图2a)。
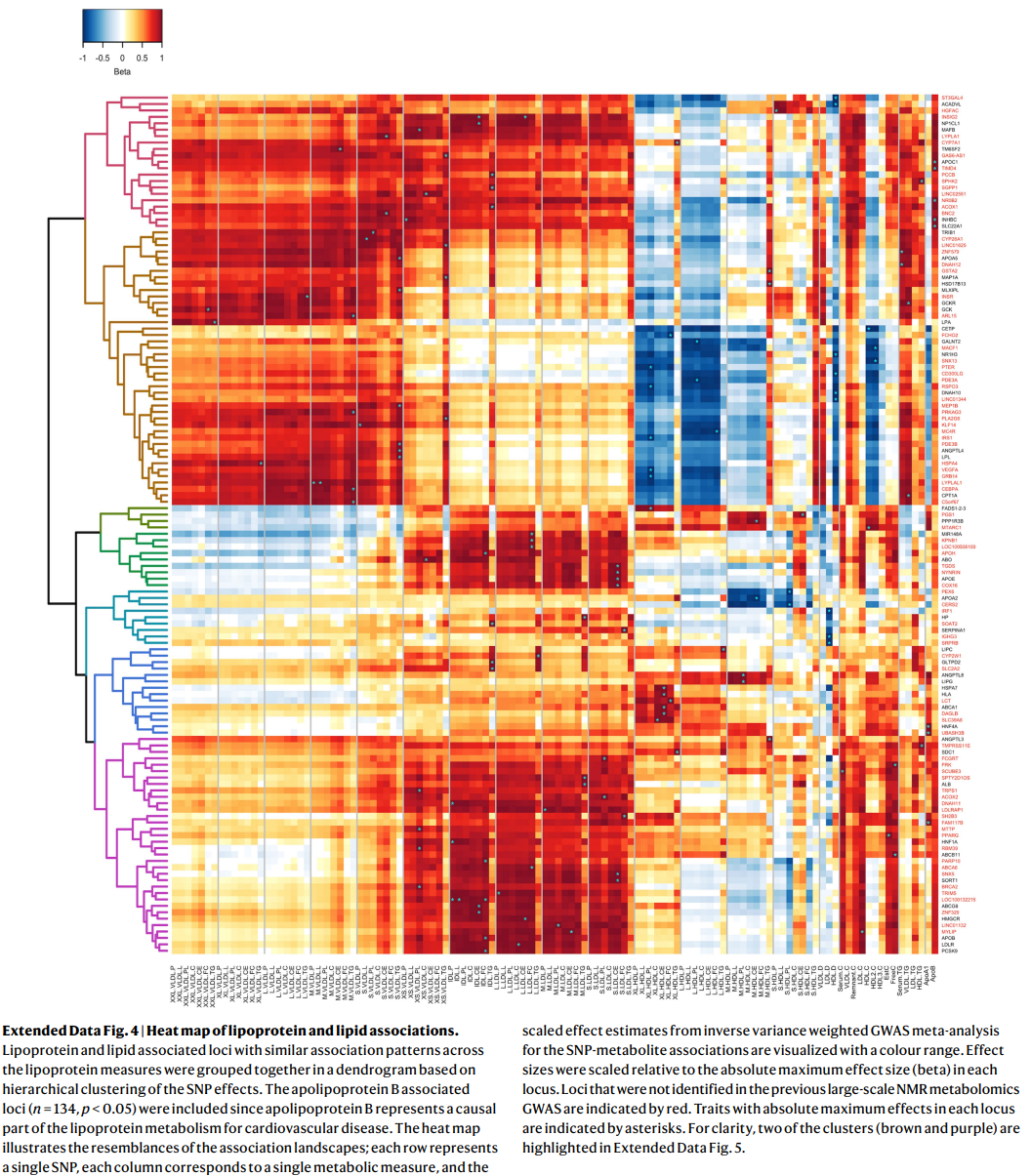
扩展数据图4 | 脂蛋白和脂质关联的热图。
具有相似关联模式的脂蛋白和脂质相关位点被分组在一起,根据SNP效应的分层聚类形成一个树状图。因为载脂蛋白B代表心血管疾病脂质代谢的一个因果部分,所以载脂蛋白B相关位点(n=134,p<0.05)被包括在内。热图展示了关联景观的相似性;每一行代表一个单一的SNP,每一列对应一个单一的代谢测量,并且通过一个颜色范围来可视化SNP-代谢物关联的逆方差加权GWAS meta分析的标准化效应估计。效应大小相对于每个位点中的绝对最大效应大小(beta)进行了缩放。在以前的大规模NMR代谢组学GWAS中未被识别的位点用红色标示。每个位点中绝对最大效应的特征用星号表示。为了清晰起见,扩展数据图5中突出显示了两个聚类(棕色和紫色)。
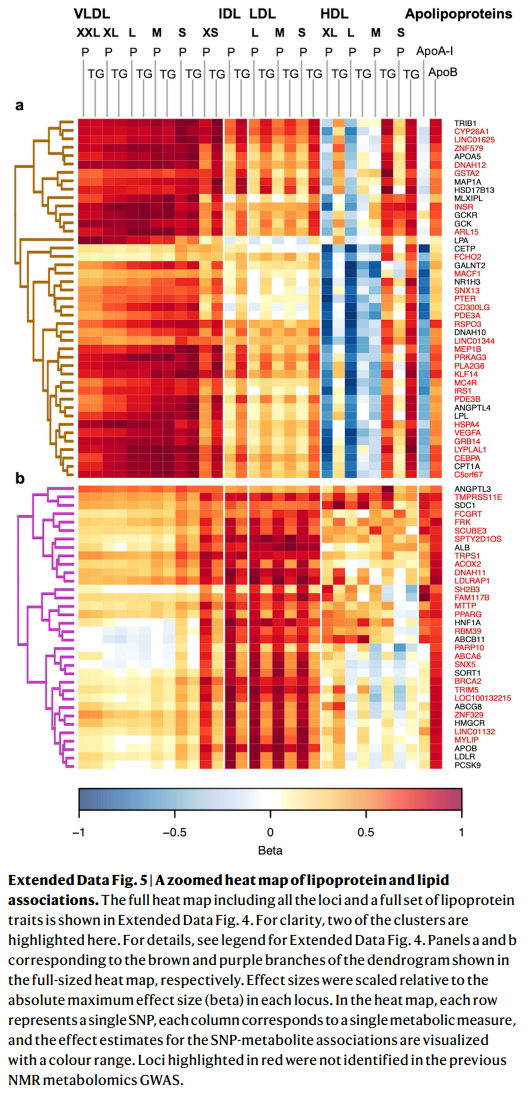
扩展数据图. 5 | 脂蛋白和脂质关联的放大热图。
完整的热图包括所有基因座和全套脂蛋白性状(见扩展数据图4)。面板a和b分别对应于完整大小热图中显示的棕色和紫色树枝。效应大小相对于每个基因座中的绝对最大效应大小(β)进行了缩放。在热图中,每一行代表一个单个SNP,每一列对应一个单个代谢测量,SNP-代谢产物关联的效应估计通过颜色范围进行可视化。以红色突出显示的基因座未在先前的NMR代谢组学GWAS中识别到。 图2 | SNPs在脂蛋白和脂质特征上的效应。 a. 脂蛋白亚类粒子浓度的相关结构热图(左)和代表性SNP的关联图景(右)。在热图中,脂蛋白亚类粒子浓度的成对相关性(在FINRISK 1997中计算;左)和SNP-代谢特征关联的效应估计(右)被表示为颜色范围。SNP效应大小相对于每个基因座中的绝对最大效应大小进行了缩放。每列代表一个单独的SNP,每行对应一个单独的代谢测量。使用双侧检验。 b,c. 效应估计(β和95%置信区间)的散点图(b)和森林图(c),跨脂蛋白和脂质特征的TRIM5和HMGCR lead snp(分别为rs11601507和rs12916;n=136,016个体)。b. 显示了最佳拟合回归线(紫色虚线)和Pearson相关系数R的估计值(对于116个SNP-特征对的beta值)。效应估计(s.d.单位)相对于LDL胆固醇减少一个s.d. 进行了缩放。VLDL,LDL,IDL和HDL分为不同的粒子大小(按大小降序排列:XXL,XL,L,M,S和XS)。代谢特征和缩写的详细描述请参见补充表2。ApoA1,载脂蛋白A1。 在此,作者使用聚类方法对先前NMR GWAS未识别的84个新位点的代谢特征进行了表征(扩展数据图4和5)。由于他们采用的方法使用了缩放效应估计,因此该研究的结果不能直接与之前使用未缩放效应估计的研究进行比较, 或者使用聚类中每种脂蛋白类型的关联数量的研究进行比较。尽管许多位点,如主调节基因PCSK9和LDLR,其聚类结果与以前的报道大致相似,但这里应用的遗传校准方法可以具体增进对不太知名的与脂质相关位点的详细代谢效应的理解,因为它们的代谢关联模式之前尚未被表征。 三联基序含蛋白5基因(TRIM5)是一个特征不明显的位点的例子,与42种脂蛋白和脂质特征相关(补充表5)。TRIM5最著名的作用是在抗病毒宿主防御中,但TRIM5附近的变异也与几个与肝脏生物学相关的特征相关,如肝酶水平,最近还报道与冠状动脉疾病的风险相关。值得注意的是,TRIM5的lead变异(rs11601507,p.Val112Ile)对脂蛋白和脂质特征的代谢影响与HMGCR的变异rs12916的代谢影响类似,后者的代谢效应与他汀类治疗一致。TRIM5如何影响脂质和脂蛋白水平及其导致冠状动脉疾病的机制尚不清楚,有猜测认为它可能与先天免疫相关。最近一项使用非酒精性脂肪肝病模型的小鼠研究表明,TRIM5可能介导DEAD-box蛋白5的降解,这可能影响mTORC1信号传导和LDL受体途径,从而影响脂质积累和炎症。无论病理生理机制如何,该研究的发现提出了一种可能性,即抑制TRIM5可以提供一种通过降低循环中含动脉粥样硬化apoB的脂蛋白颗粒的浓度,从而减少心血管疾病风险的替代治疗途径,类似于PCSK9抑制疗法,该疗法对他汀类药物不耐受者或需要进一步降低风险的他汀类药物使用者有益。虽然他们特别选择了TRIM5关联进行进一步调查,但该研究的聚类分析表明还有其他几个新位点值得进一步深入研究。 6、代谢特征变异与疾病 为了探索与代谢特征相关的变异在疾病中的作用,作者扫描了1,447个lead snp在以下两个方面的所有疾病和特征关联:(1)FinnGen研究(多达309,154名参与者,3,095种表型),这是一个将芬兰参与者的基因组信息与数字医疗数据链接的数据集;以及(2)已发布的GWAS集合,包括PhenoScanner和GWAS catalog(补充表5)。此外,还检测了SNP与基因表达(eQTL)和蛋白质水平(pQTL)的关联。大部分(1,279个)lead snp之前已报道与特征或疾病的关联(P<5×10^-8),包括直接相关的结果,如他汀类药物使用和高胆固醇血症(补充表5)。大多数SNP(1,270个)也与信使RNA(mRNA)或蛋白质水平相关(补充表5),表明至少一些关联可能是通过SNP对mRNA或蛋白质水平的直接或间接影响介导的。七个与代谢特征相关的位点(GCKR、ABCG8、ABCB11、ABCB1、CYP7A1、SERPINA1和HNF4A)与FinnGen研究中妊娠期肝内胆汁淤积症(ICP)的风险相关联(p<5×10-8)(图3a),除了ABCG8外,所有这些位点都显示出与代谢特征关联的共定位或共享区域关联的有力证据。ICP是一种在妊娠第二或第三孕期开始的胆汁淤积性疾病。 这种疾病的特征是瘙痒和血清氨基转移酶及胆汁酸浓度升高。ICP增加了羊水中胎便染色、早产、胎儿心动过缓、胎儿窘迫和胎儿丧失的风险。ICP的遗传背景尚不清楚,已发布的GWAS非常少,并且尚未表征ICP位点的代谢效应。与最近一项包括早期FinnGen发布数据和其他两个队列的meta分析数据的ICP GWAS的结果相比,在九个位点(GCKR、ABCG8、ABCB11、ABCB1–ABCB4、CYP7A1、SERPINA1、GAPDHS–TMEM147、SULT2A1和HNF4A)的关联在此得到了复制,并且另外识别了三个新位点(UGT8、NUP153和HKDC1)。在两个位点的ABCB11和ABCB4基因中,罕见的编码变异已经在ICP中被先前报道。ICP相关位点的通路分析显示,与代谢特征关联一致的是,与胆汁酸、葡萄糖和脂质代谢相关的生物过程在ICP中富集。对于某些位点(CYP7A1、ABCB1和SERPINA1),最显著的关联是检测到IDL和LDL颗粒,而两个位点(HNF4A和GCKR)更具多效性,影响范围涵盖apoB含量颗粒和HDL颗粒(图3b)。在三个位点(CYP7A1、ABCB1和SERPINA1)中,ICP易感等位基因与IDL和LDL亚类测量的浓度增高相关,而其他位点(GCKR、ABCB11和HNF4A)的关联方向则相反。当考虑将这些基因作为治疗靶点时,这些信息可能有用,因为在孕妇中不利地影响动脉粥样硬化脂质的靶点可能是不受欢迎的,尽管治疗期相对较短。通过详细表征ICP相关位点与代谢特征的关联,该研究展示了将代谢关联信息与疾病关联结合起来的价值,以阐明难以理解病状的代谢基础。 图3 | 代谢特征相关变异与ICP相关。 a,b. 妊娠期肝内胆汁淤积(ICP)GWAS的曼哈顿图(a)和与代谢特征和ICP相关的基因座热图(b)。在FinnGen研究中,12个基因座与ICP相关(1,460例病例,172,286例对照)。a. 突显了紧邻lead snp的500 kb区域,并指示了每个信号的最近基因。ICP GWAS采用广义混合模型(SAIGE)进行。指示了与NMR meta分析中鉴定的基因座重叠的基因座。b. 包括可能与代谢特征具有共享致因变异的基因座。热图展示了关联景观的相似性。每行代表一个单独的SNP,每列对应一个单独的代谢测量,来自逆方差加权的GWAS meta分析的SNP-代谢特征关联的缩放效应估计表示为颜色范围。 7、孟德尔随机化 最终,作者利用GWAS meta分析中没有包括UK Biobank的情况,进行了两样本孟德尔随机化分析,以探索遗传预测的20种非脂质特征与UK Biobank中460个Phecode和52个定量特征的关联。使用每个特征的所有lead变异作为遗传工具进行的初步孟德尔随机化分析,在逆方差加权模型下识别出503个显著关联(p<4.88×10-6),包括葡萄糖与糖尿病、肌酐与肾衰竭以及氨基酸与糖尿病之间的正相关关系,所有这些都代表了已知的因果关系。较少特征化的关系包括遗传预测的乳酸水平与子宫良性肿瘤之间的正相关。这种潜在的因果关系与最近的一项GWAS相符,该研究将遗传倾向于提高肌肉质量与子宫纤维瘤联系起来。他们还发现遗传预测的血液中甘氨酸水平与血压之间存在反向关联,这一发现得到了其与高血压的强烈观察性关联和遗传数据的支持。这一发现表明甘氨酸可能是先前报道的甘氨酸水平与心肌梗死反向关联的潜在中介。 这些例子强调了将遗传学、代谢特征和疾病结果的数据联系起来的价值,以大规模识别代谢特征与疾病之间的新因果关系。将分析限制在与少于5种代谢特征相关的较少多效性变异上时,关联估计平均明显较弱,变异间异质性较小(中位绝对beta值,0.058 vs.0.152;Q统计量,34.2 vs.385.6),表明多效性可能是驱动初步孟德尔随机化关联的因素。使用两个不同的变异多效性阈值(与少于三个和少于七个代谢特征相关的多效性SNP)的结果非常相似,表明发现不受阈值选择的影响。这明确强调在选择用于孟德尔随机化的工具SNP时应仔细考虑多效性,以避免对潜在因果关系的错误解释。 作为一个例子,乙酮的孟德尔随机化结果受到包含更多的多效性SNP工具影响显著(图4)。乙酮是一种主要在禁食时肝脏产生的酮体,已在生化和流行病学研究中与多种心脏代谢状况相关,包括心力衰竭和糖尿病。在GWAS中,他们在十个位点发现了乙酮的关联(之前的NMR GWAS meta分析中只识别到一个相关位点—APOA5),孟德尔随机化产生了20个稳健的关联(图4a)。这些包括与甘油三酯、高密度脂蛋白胆固醇和残余胆固醇的关联,这可能反映了包括已知脂质位点(LPL、APOA5、TRIB1、APOC1、GALNT2和PPP1R3B)在内的工具。乙酮的较少多效性工具仅包括四个位点:3-羟基-3-甲基戊二酸辅酶A合成酶2(HMGCS2)、3-酮酸辅酶A转移酶1(OXCT1)、细胞色素P450家族2亚家族E成员1(CYP2E1)和SLC2A4,所有这些都在酮体或血糖相关途径中具有直接作用。仅使用这4个变异体,与高血压的正相关关联(每标准差更高的遗传预测乙酮水平的风险比=1.41,P=6.9×10^-7)是稳健的(图4a、b),并在FinnGen中也得到复制(风险比1.45,P=4.5×10^-5)(图4c)。与这些结果一致,乙酮最近被建议作为高血压的生物标志物。应该注意的是,先前使用NMR代谢组学平台的研究错误地将乙酮标记为乙酰乙酸,这一错误在2020年及之后版本的平台中得到了纠正。乙酮与高血压之间潜在因果关系的发现值得注意,因为关于酮酸饮食在高血压中的作用的数据虽有启示但不确定,并且酮体也已成为冠状动脉疾病潜在的治疗剂。这一发现与当前临床和人类研究相符,这些研究将改变酮体水平的干预措施,如酮酸饮食和酮盐补充,与血压变化联系起来,从而提出酮体可能是高血压和其他心血管疾病的有前途的潜在治疗策略。酮体影响高血压风险的机制目前尚不清楚,提出了直接(例如,交感神经系统活动、血管舒张和心脏内皮细胞增殖)和间接(例如,肥胖和糖尿病)的途径。最近在UK Biobank的一项研究显示,与非脂质NMR特征相关的一些位点和途径高度多效性,较少多效性变异体往往更能反映与特征更接近的生物学过程。这也符合作者的发现,通过识别几个与甘油三酯相关的多效性基因与乙酮水平相关,以及四个与酮体生物学途径直接相关的较少多效性乙酮相关位点。这些结果强调了代谢测量的遗传多效性很常见,即使是对于某些非脂质特征也是如此,并且在孟德尔随机化中仔细选择变异体至关重要,以避免由于普遍存在的多效性而造成的偏见。 这些研究结果突出了孟德尔随机化分析在揭示代谢特征与疾病之间的潜在因果关系中的应用价值,特别是当这些关系通过传统的流行病学方法难以证明时。通过使用遗传变异作为工具变量,这种方法可以帮助理解特定基因变异如何通过影响代谢途径间接或直接影响疾病风险,从而为发展新的治疗策略提供科学依据。此外,这种方法还能揭示哪些生物标志物可能在疾病预防和管理中起重要作用,进一步指导公共卫生策略和个体化医疗的实施。 最终,孟德尔随机化的发展和应用提醒我们,在进行这类分析时需谨慎考虑遗传多效性的潜在影响,以确保结果的准确性和可靠性。这要求研究者在设计研究时选择适当的统计模型和分析方法,以及在解释结果时充分考虑生物学背景和路径的复杂性。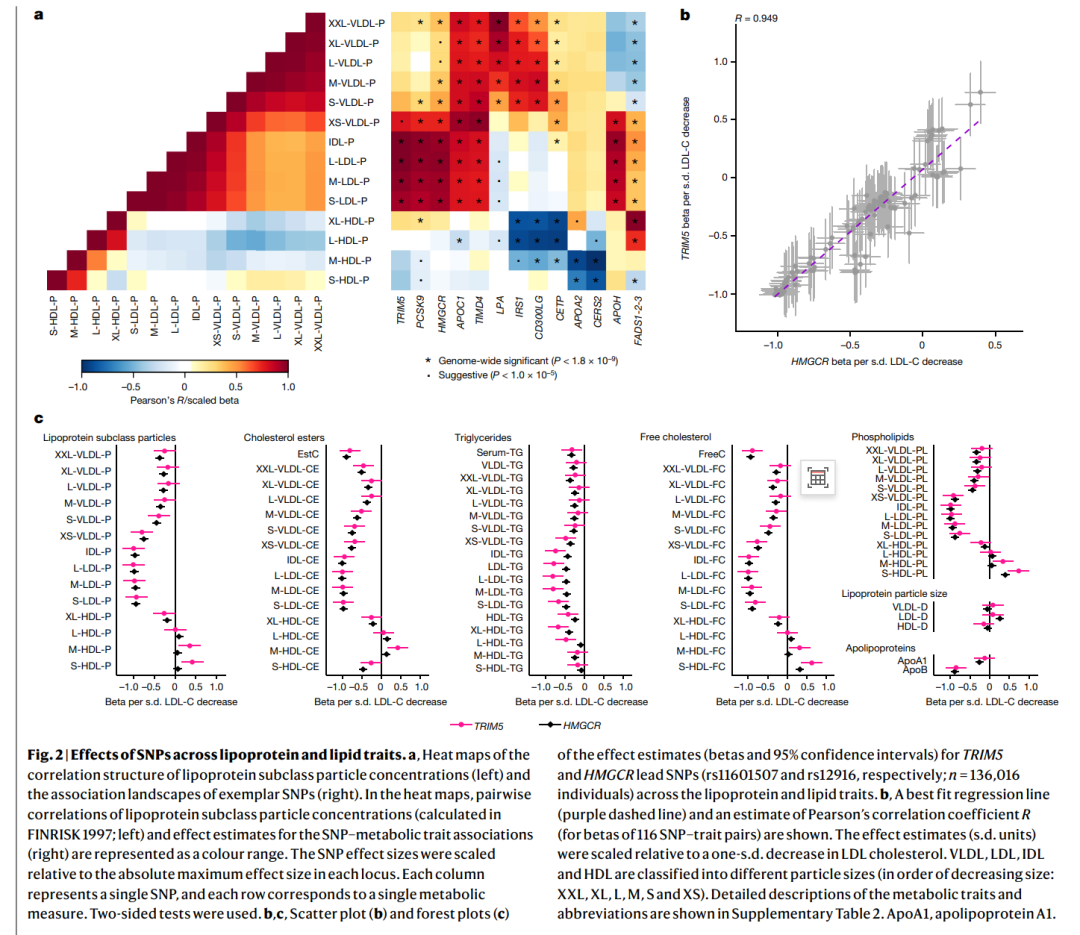
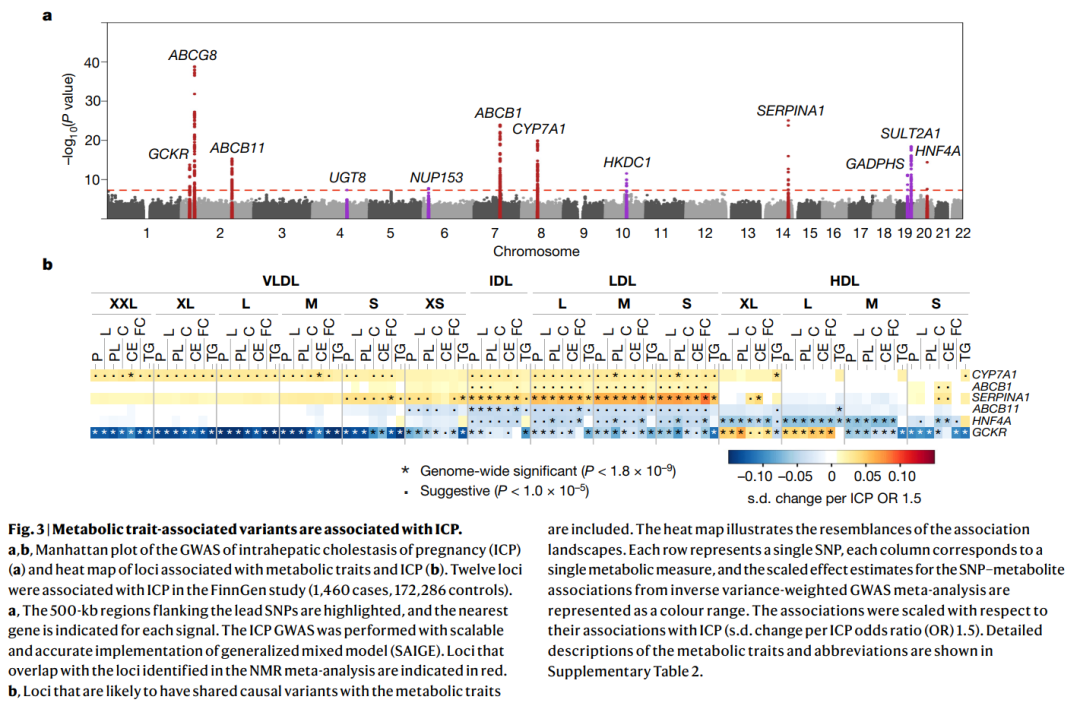
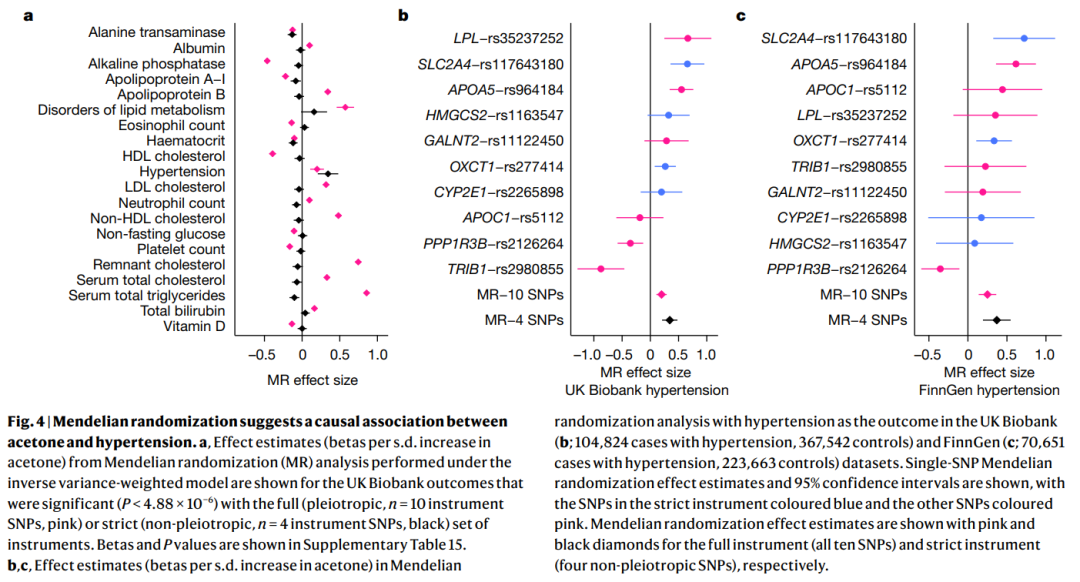
图4 | 遗传随机分析表明丙酮与高血压之间存在因果关系。
a. 在逆方差加权模型下进行的遗传随机分析(MR)中显示了对UK Biobank结果的效应估计(每个丙酮标准差增加的β),这些结果是与全(多功能,n=10工具SNP,粉色)或严格(非多功能,n=4工具SNP,黑色)工具组合显著的(p<4.88×10−6)。效应估计和P值在补充表15中显示。b,c. 在UK Biobank(b;104,824例高血压病例,367,542例对照)和FinnGen(c;70,651例高血压病例,223,663例对照)数据集中以高血压为结果的遗传随机分析中的效应估计(每个丙酮标准差增加的β)。显示了单个SNP遗传随机效应估计和95%置信区间,严格工具中的SNP为蓝色,其他SNP为粉色。遗传随机效应估计用粉色和黑色钻石表示全工具(所有十个SNP)和严格工具(四个非多功能SNP)。 讨论: 1、局限性 参与者主要是欧洲血统(27个队列中的33个),这意味着作者检测其他血统群体关联的能力有限。然而,作者按血统分层比较表明,所发现的关联在不同血统间具有较广泛的可转移性。未来需要进行更大规模的多血统研究,包括非洲血统,以更好地理解全球范围内的代谢遗传调控。该研究基于NMR的研究在分析的代谢特征数量上也受到限制,与使用质谱法的研究相比较少,后者是一种可以同时测量成千上万种代谢物的互补方法。尽管质谱法更为敏感,NMR在分析上更为稳健、高通量且成本低,因此该研究包括的参与者数量是基于质谱的最大GWAS研究的六倍多,这使得遗传调控的特征能被更深入地表征。此外,质谱法无法提供NMR平台能够提供的脂蛋白亚类的详细分析。另一个局限是,尽管作者根据禁食状态和样本类型识别了遗传关联的差异,但解释这些差异的机制仍然只是推测性的,需要进一步研究。这些差异表明,在解释不同研究中的遗传力估计时应谨慎,例如UK Biobank。此外,作者描述了与ICP相关的遗传位点的详细代谢关联,应注意许多与ICP相关的位点已知与肝功能酶或胆红素相关,这些水平的增加被纳入ICP的诊断标准中。然而,诊断ICP需要有瘙痒的出现,通过GWAS包括的医院出院登记定义的ICP病例因此应代表真实的有症状病例。 2、结论 通过这项涉及超过136,000名参与者的大规模全基因组meta分析,该研究识别了超过8,000个与循环代谢生物标志物相关的遗传关联,涉及400多个基因座。与之前的NMR代谢特征的GWAS meta分析相比,样本量增加了五倍,代谢特征的数量增加了一倍,显著增加了显著关联的数量(之前有62个相关位点),从而大幅提高了对系统代谢遗传调控的理解。该研究meta分析的关键特点是包括了33个队列的参与者,这使作者能够发现许多来自独立数据集的新的稳健关联。通过这些数据集的内部比较以及与UK Biobank的外部比较,该研究强调了样本和参与者特征(如样本类型和禁食状态)在揭示或掩盖遗传关联中的重要作用,这对生物学解释和后续分析有重要影响。 作者对近300个相关位点进行了广泛的手动整理,以识别高度可能的因果基因,这为进一步理解这些关联提供了有用的资源,并允许高信度地识别与之共定位的疾病关联的因果基因。对于其余的位点,该研究的结果为识别到目前为止尚未知涉及代谢调控的基因提供了一个起点。此外,其对脂蛋白测量的细粒度代谢关联的比较使该研究能够识别具有相似代谢特征的基因簇,这表明TRIM5是一个潜在的治疗靶点,用于降低促动脉粥样硬化的脂质水平,因此也用于心血管疾病,因为TRIM5的代谢特征与通过LDL受体影响肝细胞LDL胆固醇摄取的基因相吻合。通过公开汇总统计数据,该研究为孟德尔随机化研究提供了宝贵的资源,并说明了使用多效性变异作为遗传工具变量的潜在陷阱。最后,作者展示了利用这些发现来阐明病因尚不明确的疾病的潜力。 汇报人: 王肖宇 导师:赵宇 审核:张子妍 任建君
